

Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis
- PMID: 21820099
- PMCID: PMC3155175
- DOI: 10.1016/j.ajhg.2011.07.003
Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis
Erratum in
- Am J Hum Genet. 2011 Oct 7;89(4):589
Abstract
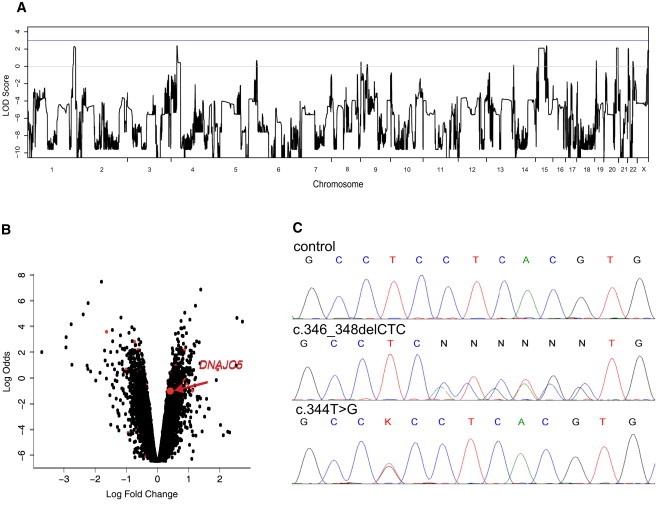
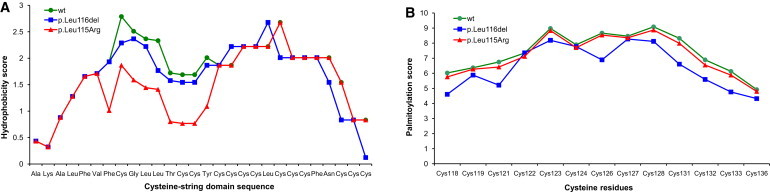
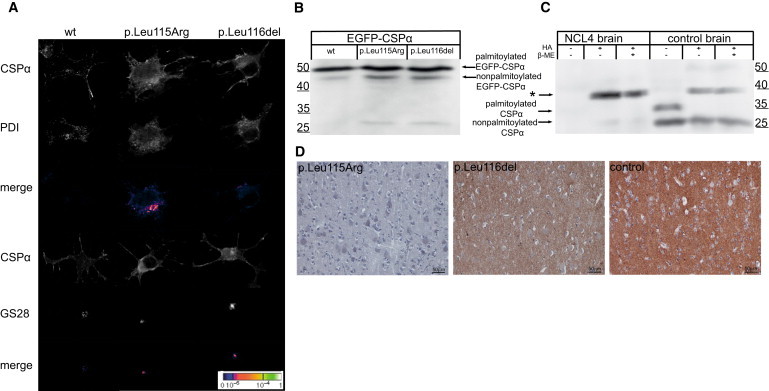
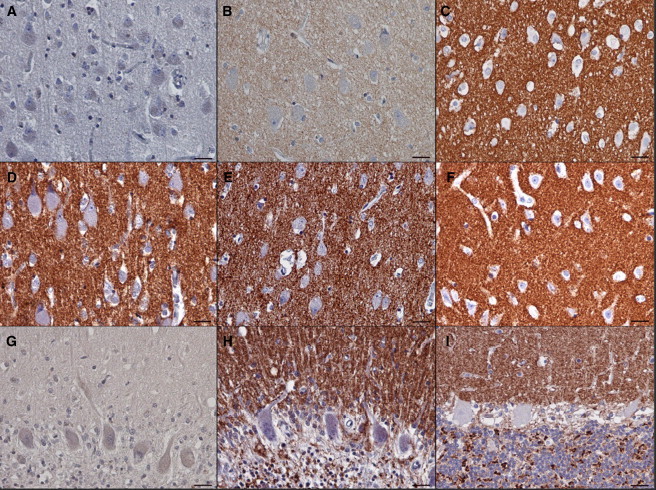
Autosomal-dominant adult-onset neuronal ceroid lipofuscinosis (ANCL) is characterized by accumulation of autofluorescent storage material in neural tissues and neurodegeneration and has an age of onset in the third decade of life or later. The genetic and molecular basis of the disease has remained unknown for many years. We carried out linkage mapping, gene-expression analysis, exome sequencing, and candidate-gene sequencing in affected individuals from 20 families and/or individuals with simplex cases; we identified in five individuals one of two disease-causing mutations, c.346_348delCTC and c.344T>G, in DNAJC5 encoding cysteine-string protein alpha (CSPα). These mutations-causing a deletion, p.Leu116del, and an amino acid exchange, p.Leu115Arg, respectively-are located within the cysteine-string domain of the protein and affect both palmitoylation-dependent sorting and the amount of CSPα in neuronal cells. The resulting depletion of functional CSPα might cause in parallel the presynaptic dysfunction and the progressive neurodegeneration observed in affected individuals and lysosomal accumulation of misfolded and proteolysis-resistant proteins in the form of characteristic ceroid deposits in neurons. Our work represents an important step in the genetic dissection of a genetically heterogeneous group of ANCLs. It also confirms a neuroprotective role for CSPα in humans and demonstrates the need for detailed investigation of CSPα in the neuronal ceroid lipofuscinoses and other neurodegenerative diseases presenting with neuronal protein aggregation.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Mole S.E., Williams R.E., Goebel H.H. Oxford University Press; Oxford: 2011. The Neuronal Ceroid Lipofuscinoses (Batten Disease)
-
- Boehme D.H., Cottrell J.C., Leonberg S.C., Zeman W. A dominant form of neuronal ceroid-lipofuscinosis. Brain. 1971;94:745–760. - PubMed
-
- Ferrer I., Arbizu T., Peña J., Serra J.P. A golgi and ultrastructural study of a dominant form of Kufs' disease. J. Neurol. 1980;222:183–190. - PubMed
-
- Josephson S.A., Schmidt R.E., Millsap P., McManus D.Q., Morris J.C. Autosomal dominant Kufs' disease: A cause of early onset dementia. J. Neurol. Sci. 2001;188:51–60. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
