

Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease
- PMID: 21820100
- PMCID: PMC3155164
- DOI: 10.1016/j.ajhg.2011.07.002
Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease
Abstract
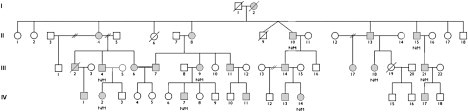
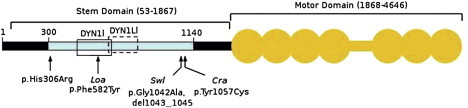
Charcot-Marie-Tooth disease is characterized by length-dependent axonal degeneration with distal sensory loss and weakness, deep-tendon-reflex abnormalities, and skeletal deformities. It is caused by mutations in more than 40 genes. We investigated a four-generation family with 23 members affected by the axonal form (type 2), for which the common causes had been excluded by Sanger sequencing. Exome sequencing of three affected individuals separated by eight meioses identified a single shared novel heterozygous variant, c.917A>G, in DYNC1H1, which encodes the cytoplasmic dynein heavy chain 1 (here, novel refers to a variant that has not been seen in dbSNP131or the August 2010 release of the 1000 Genomes project). Testing of six additional affected family members showed cosegregation and a maximum LOD score of 3.6. The shared DYNC1H1 gene variant is a missense substitution, p.His306Arg, at a highly conserved residue within the homodimerization domain. Three mouse models with different mutations within this domain have previously been reported with age-related progressive loss of muscle bulk and locomotor ability. Cytoplasmic dynein is a large multisubunit motor protein complex and has a key role in retrograde axonal transport in neurons. Our results highlight the importance of dynein and retrograde axonal transport in neuronal function in humans.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Hafezparast M., Klocke R., Ruhrberg C., Marquardt A., Ahmad-Annuar A., Bowen S., Lalli G., Witherden A.S., Hummerich H., Nicholson S. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
