

The Phenix software for automated determination of macromolecular structures
- PMID: 21821126
- PMCID: PMC3193589
- DOI: 10.1016/j.ymeth.2011.07.005
The Phenix software for automated determination of macromolecular structures
Abstract
X-ray crystallography is a critical tool in the study of biological systems. It is able to provide information that has been a prerequisite to understanding the fundamentals of life. It is also a method that is central to the development of new therapeutics for human disease. Significant time and effort are required to determine and optimize many macromolecular structures because of the need for manual interpretation of complex numerical data, often using many different software packages, and the repeated use of interactive three-dimensional graphics. The Phenix software package has been developed to provide a comprehensive system for macromolecular crystallographic structure solution with an emphasis on automation. This has required the development of new algorithms that minimize or eliminate subjective input in favor of built-in expert-systems knowledge, the automation of procedures that are traditionally performed by hand, and the development of a computational framework that allows a tight integration between the algorithms. The application of automated methods is particularly appropriate in the field of structural proteomics, where high throughput is desired. Features in Phenix for the automation of experimental phasing with subsequent model building, molecular replacement, structure refinement and validation are described and examples given of running Phenix from both the command line and graphical user interface.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
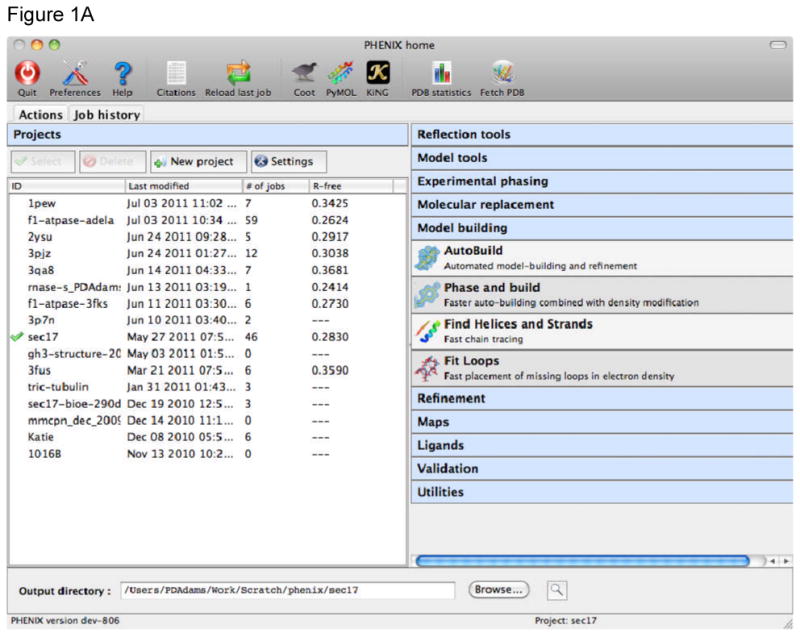
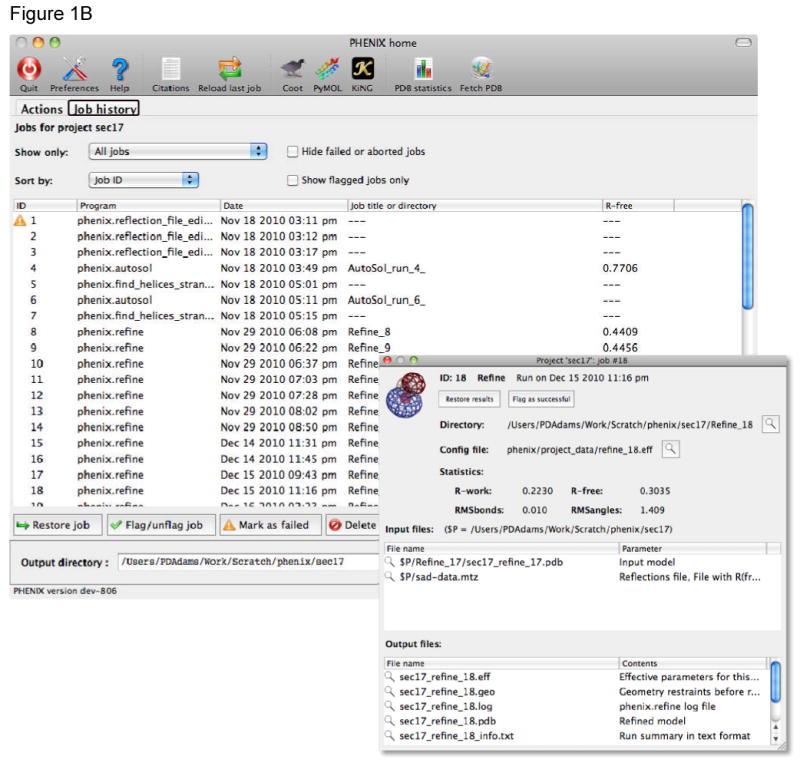
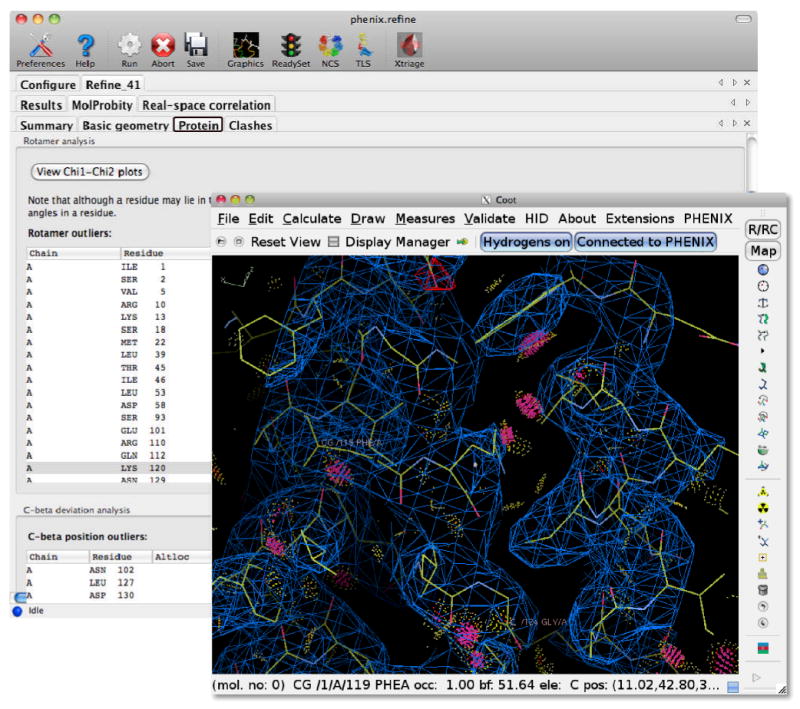
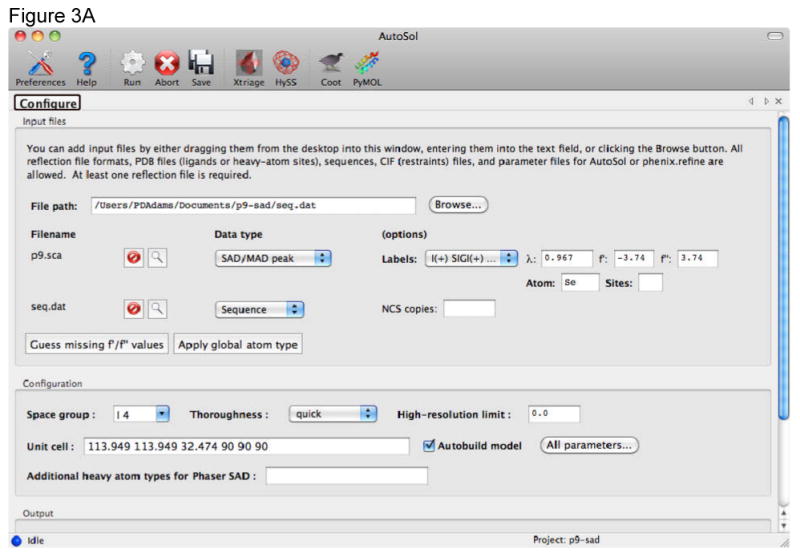
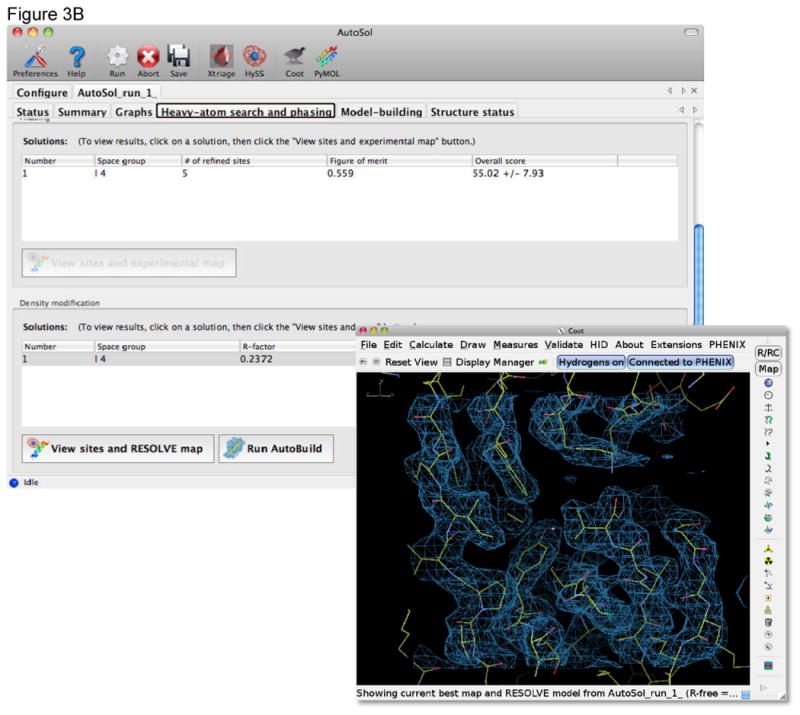
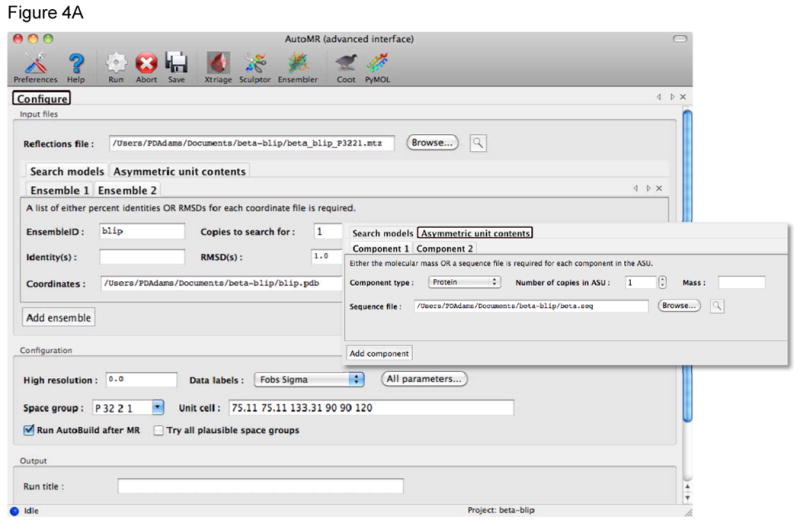
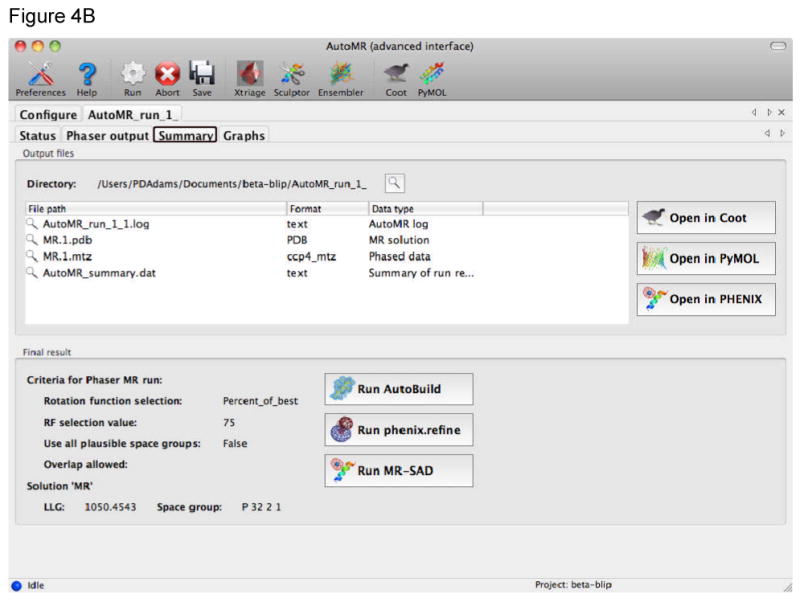
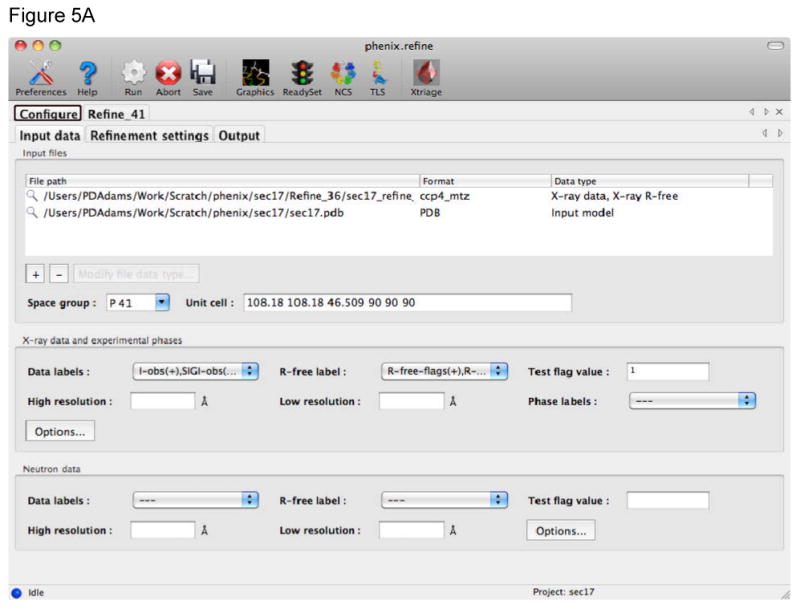
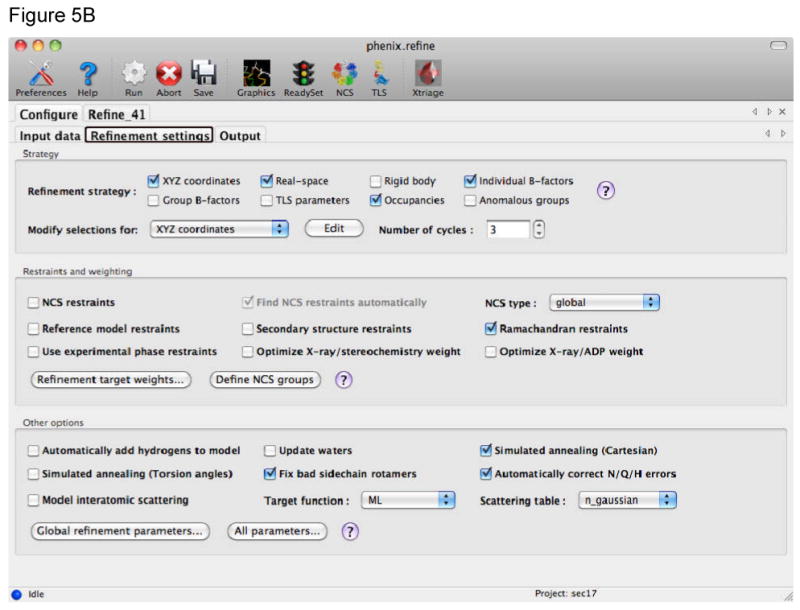
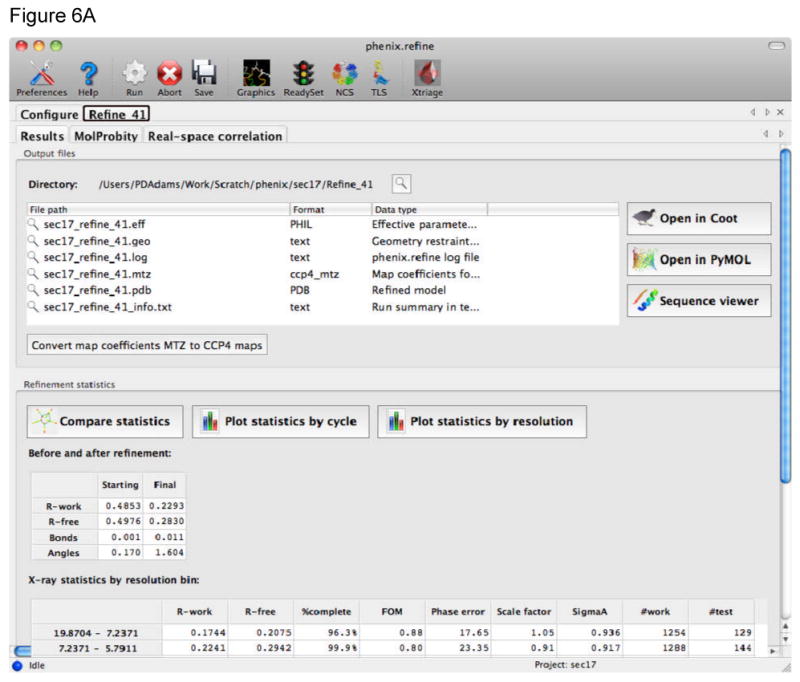
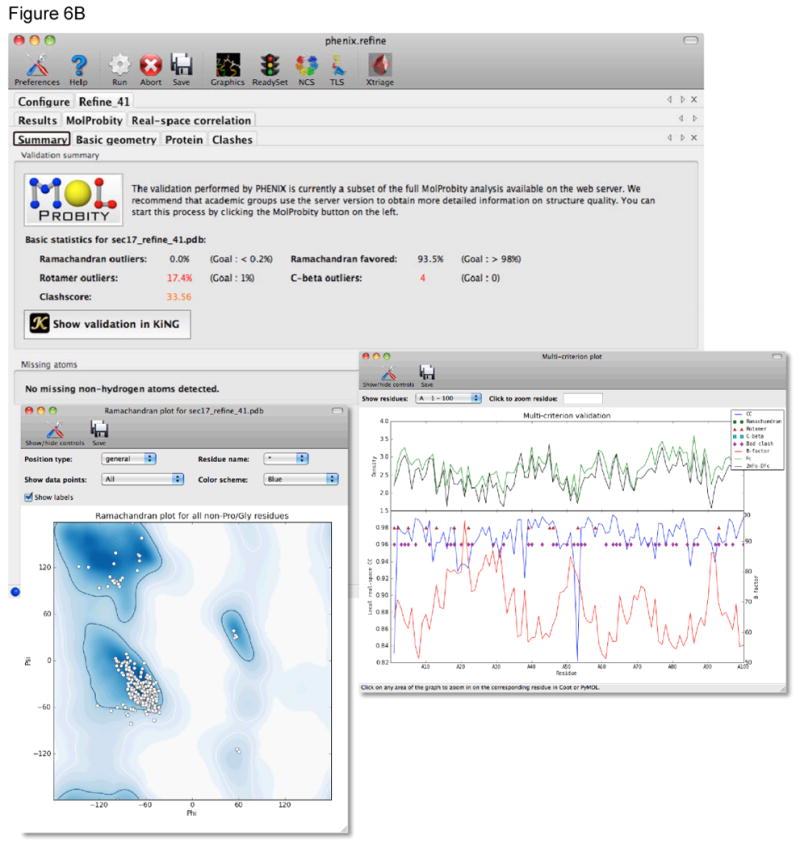
References
-
- Sheldrick GM. A short history of SHELX. Acta crystallographica Section A, Foundations of crystallography. 2008;64(Pt 1):112–22. - PubMed
-
- Allen FH, Kennard O, Taylor R. Systematic Analysis of Structural Data as a Research Technique in Organic Chemistry. Acc Chem Res. 1983;16:146–153.
-
- Weeks CM, et al. Automatic solution of heavy-atom substructures. Methods Enzymol. 2003;374:37–83. - PubMed
-
- Fortelle Edl, Bricogne G. Methods in Enzymology. Academic Press; San Diego: 1997. Maximum-Likelihood Heavy-Atom Parameter Refinement for Multiple Isomorphous Replacement and Multiwavelength Anomalous Diffraction Methods; pp. 472–494. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
