

Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival
- PMID: 21821917
- PMCID: PMC3163943
- DOI: 10.1172/JCI43382
Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival
Abstract
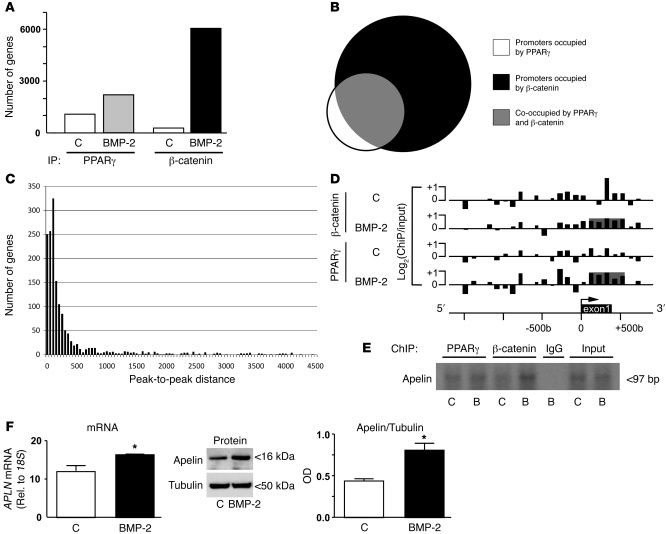
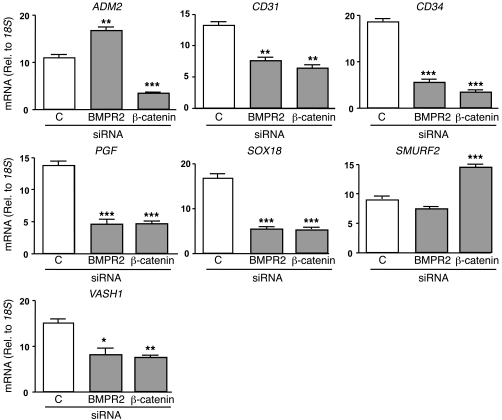
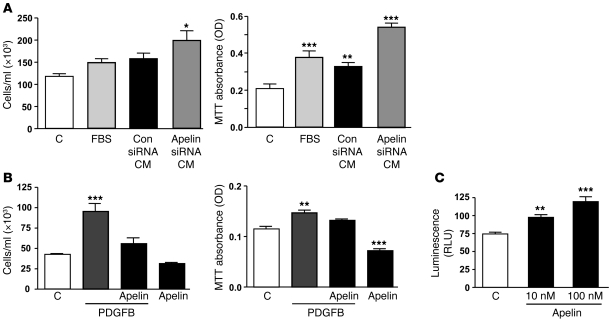
Reduced bone morphogenetic protein receptor 2 (BMPR2) expression in patients with pulmonary arterial hypertension (PAH) can impair pulmonary arterial EC (PAEC) function. This can adversely affect EC survival and promote SMC proliferation. We hypothesized that interventions to normalize expression of genes that are targets of BMPR2 signaling could restore PAEC function and prevent or reverse PAH. Here we have characterized, in human PAECs, a BMPR2-mediated transcriptional complex between PPARγ and β-catenin and shown that disruption of this complex impaired BMP-mediated PAEC survival. Using whole genome-wide ChIP-Chip promoter analysis and gene expression microarrays, we delineated PPARγ/β-catenin-dependent transcription of target genes including APLN, which encodes apelin. We documented reduced PAEC expression of apelin in PAH patients versus controls. In cell culture experiments, we showed that apelin-deficient PAECs were prone to apoptosis and promoted pulmonary arterial SMC (PASMC) proliferation. Conversely, we established that apelin, like BMPR2 ligands, suppressed proliferation and induced apoptosis of PASMCs. Consistent with these functions, administration of apelin reversed PAH in mice with reduced production of apelin resulting from deletion of PPARγ in ECs. Taken together, our findings suggest that apelin could be effective in treating PAH by rescuing BMPR2 and PAEC dysfunction.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL132550/HL/NHLBI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01-HL087118/HL/NHLBI NIH HHS/United States
- UL1RR024986/RR/NCRR NIH HHS/United States
- HL58115/HL/NHLBI NIH HHS/United States
- UL1 RR024986/RR/NCRR NIH HHS/United States
- R37 HL058115/HL/NHLBI NIH HHS/United States
- R01 HL058115/HL/NHLBI NIH HHS/United States
- R01 HL087118/HL/NHLBI NIH HHS/United States
- HL64937/HL/NHLBI NIH HHS/United States
- P01 HL108797/HL/NHLBI NIH HHS/United States
- R01 HL064937/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
