

Adapting Poisson-Boltzmann to the self-consistent mean field theory: application to protein side-chain modeling
- PMID: 21823735
- PMCID: PMC3162615
- DOI: 10.1063/1.3621831
Adapting Poisson-Boltzmann to the self-consistent mean field theory: application to protein side-chain modeling
Abstract
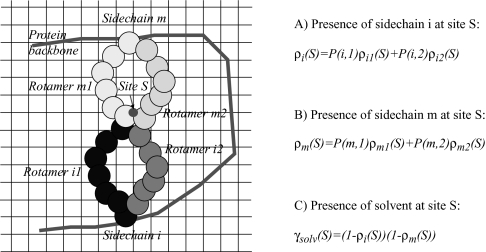
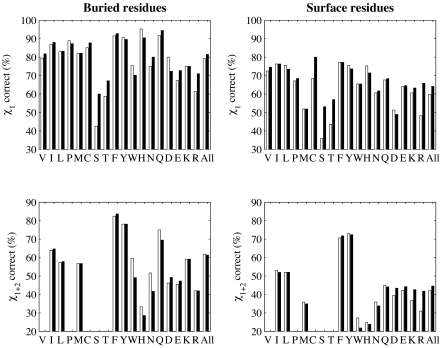
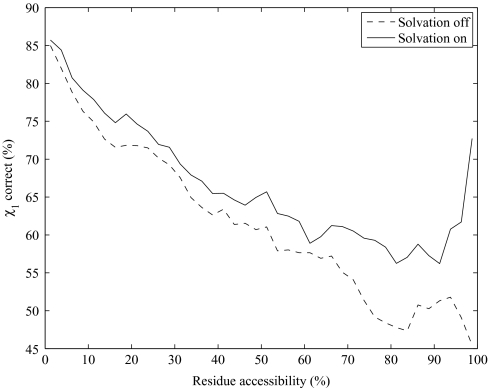
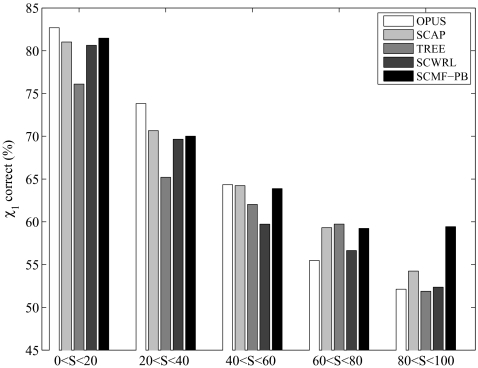
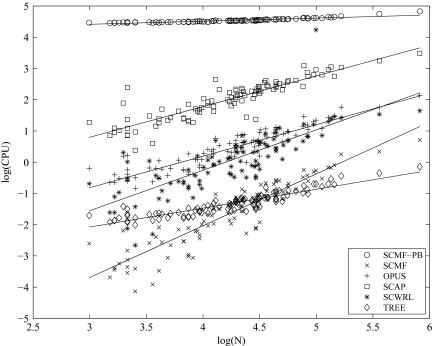
We present an extension of the self-consistent mean field theory for protein side-chain modeling in which solvation effects are included based on the Poisson-Boltzmann (PB) theory. In this approach, the protein is represented with multiple copies of its side chains. Each copy is assigned a weight that is refined iteratively based on the mean field energy generated by the rest of the protein, until self-consistency is reached. At each cycle, the variational free energy of the multi-copy system is computed; this free energy includes the internal energy of the protein that accounts for vdW and electrostatics interactions and a solvation free energy term that is computed using the PB equation. The method converges in only a few cycles and takes only minutes of central processing unit time on a commodity personal computer. The predicted conformation of each residue is then set to be its copy with the highest weight after convergence. We have tested this method on a database of hundred highly refined NMR structures to circumvent the problems of crystal packing inherent to x-ray structures. The use of the PB-derived solvation free energy significantly improves prediction accuracy for surface side chains. For example, the prediction accuracies for χ(1) for surface cysteine, serine, and threonine residues improve from 68%, 35%, and 43% to 80%, 53%, and 57%, respectively. A comparison with other side-chain prediction algorithms demonstrates that our approach is consistently better in predicting the conformations of exposed side chains.
Figures
References
-
- Chazelle B., Kinsfort C., and Singh M., INFORMS J. Comput. 16, 380 (2004).10.1287/ijoc.1040.0096 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources