

Understanding the impact of 1q21.1 copy number variant
- PMID: 21824431
- PMCID: PMC3180300
- DOI: 10.1186/1750-1172-6-54
Understanding the impact of 1q21.1 copy number variant
Abstract
Background: 1q21.1 Copy Number Variant (CNV) is associated with a highly variable phenotype ranging from congenital anomalies, learning deficits/intellectual disability (ID), to a normal phenotype. Hence, the clinical significance of this CNV can be difficult to evaluate. Here we described the consequences of the 1q21.1 CNV on genome-wide gene expression and function of selected candidate genes within 1q21.1 using cell lines from clinically well described subjects.
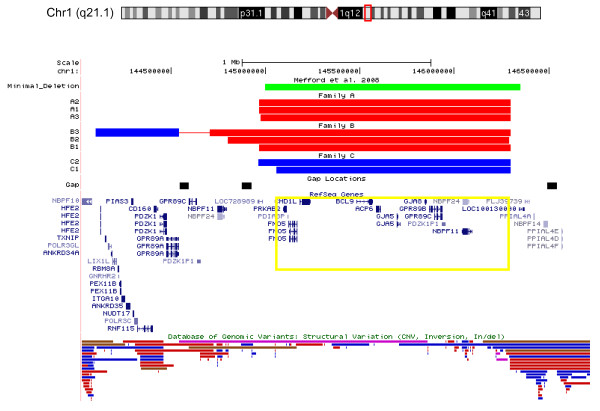
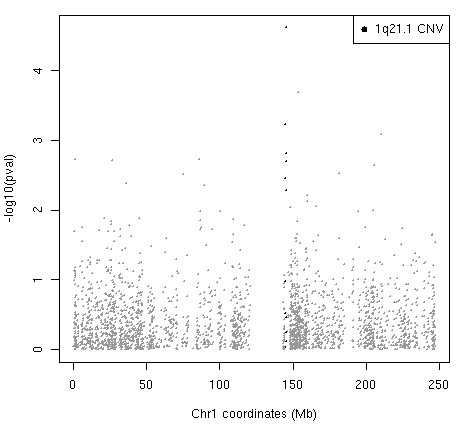
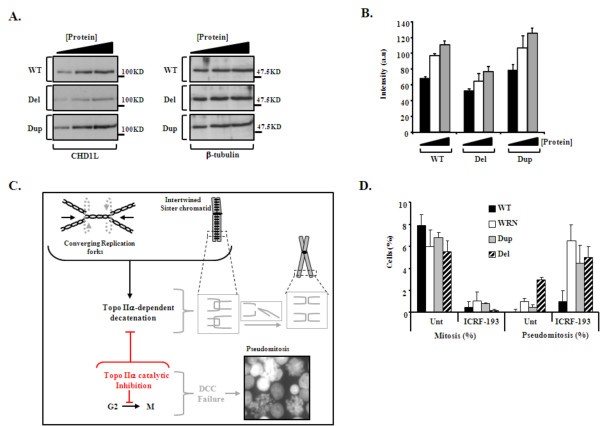
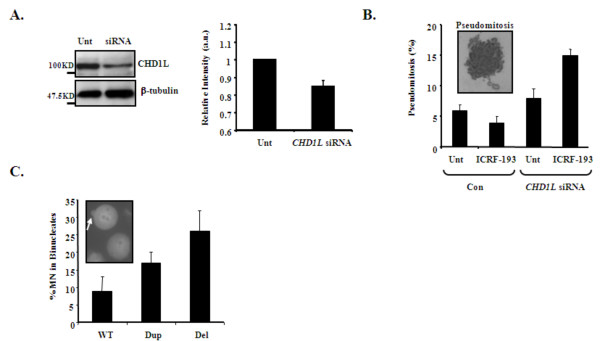
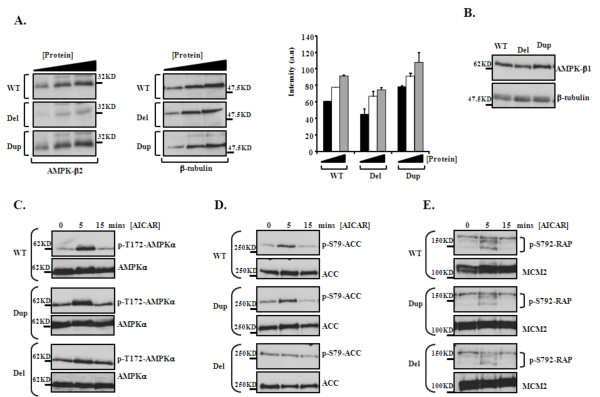
Methods and results: Eight subjects from 3 families were included in the study: six with a 1q21.1 deletion and two with a 1q21.1 duplication. High resolution Affymetrix 2.7M array was used to refine the 1q21.1 CNV breakpoints and exclude the presence of secondary CNVs of pathogenic relevance. Whole genome expression profiling, studied in lymphoblast cell lines (LBCs) from 5 subjects, showed enrichment of genes from 1q21.1 in the top 100 genes ranked based on correlation of expression with 1q21.1 copy number. The function of two top genes from 1q21.1, CHD1L/ALC1 and PRKAB2, was studied in detail in LBCs from a deletion and a duplication carrier. CHD1L/ALC1 is an enzyme with a role in chromatin modification and DNA damage response while PRKAB2 is a member of the AMP kinase complex, which senses and maintains systemic and cellular energy balance. The protein levels for CHD1L/ALC1 and PRKAB2 were changed in concordance with their copy number in both LBCs. A defect in chromatin remodeling was documented based on impaired decatenation (chromatid untangling) checkpoint (DCC) in both LBCs. This defect, reproduced by CHD1L/ALC1 siRNA, identifies a new role of CHD1L/ALC1 in DCC. Both LBCs also showed elevated levels of micronuclei following treatment with a Topoisomerase II inhibitor suggesting increased DNA breaks. AMP kinase function, specifically in the deletion containing LBCs, was attenuated.
Conclusion: Our studies are unique as they show for the first time that the 1q21.1 CNV not only causes changes in the expression of its key integral genes, associated with changes at the protein level, but also results in changes in their known function, in the case of AMPK, and newly identified function such as DCC activation in the case of CHD1L/ALC1. Our results support the use of patient lymphoblasts for dissecting the functional sequelae of genes integral to CNVs in carrier cell lines, ultimately enhancing understanding of biological processes which may contribute to the clinical phenotype.
Figures
References
-
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, Shen J, Kang SH, Pursley A, Lotze T, Kennedy G, Lansky-Shafer S, Weaver C, Roeder ER, Grebe TA, Arnold GL, Hutchison T, Reimschisel T, Amato S, Geragthy MT, Innis JW, Obersztyn E, Nowakowska B, Rosengren SS, Bader PI, Grange DK. et al.Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nature genetics. 2008;40:1466–1471. doi: 10.1038/ng.279. - DOI - PMC - PubMed
-
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV. et al.Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. The New England journal of medicine. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. - DOI - PMC - PubMed
-
- Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A. et al.Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. - DOI - PMC - PubMed
-
- Tam GW, van de Lagemaat LN, Redon R, Strathdee KE, Croning MD, Malloy MP, Muir WJ, Pickard BS, Deary IJ, Blackwood DH, Carter NP, Grant SG. Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochemical Society transactions. 2010;38:445–451. doi: 10.1042/BST0380445. - DOI - PubMed
-
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS. et al.Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science (New York, NY) 2008;320:539–543. doi: 10.1126/science.1155174. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
