

The functional cycle of visual arrestins in photoreceptor cells
- PMID: 21824527
- PMCID: PMC3196764
- DOI: 10.1016/j.preteyeres.2011.07.002
The functional cycle of visual arrestins in photoreceptor cells
Abstract
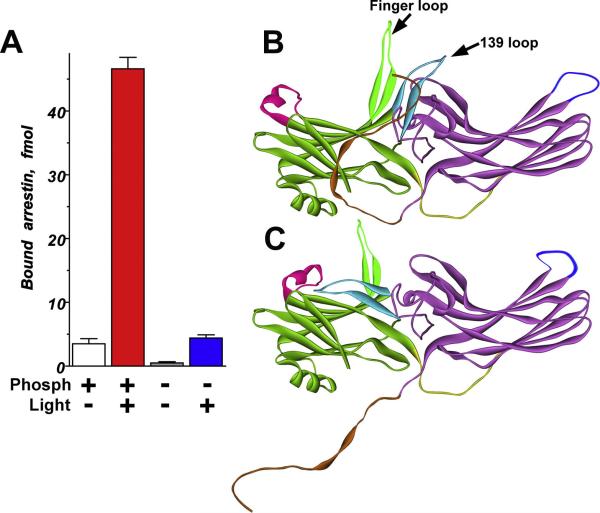
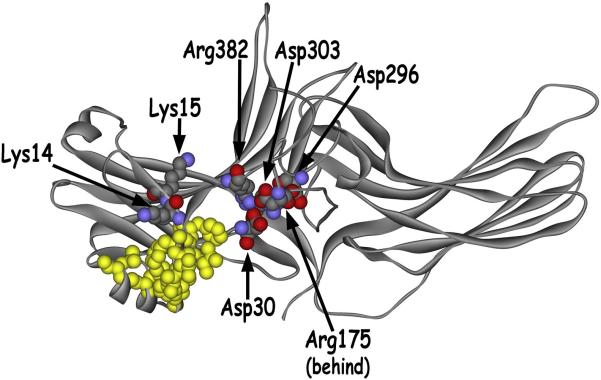
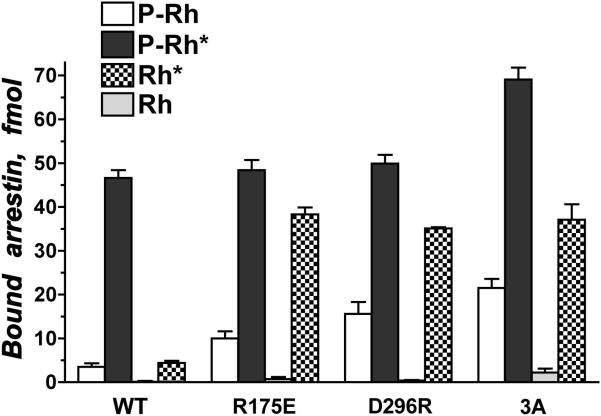
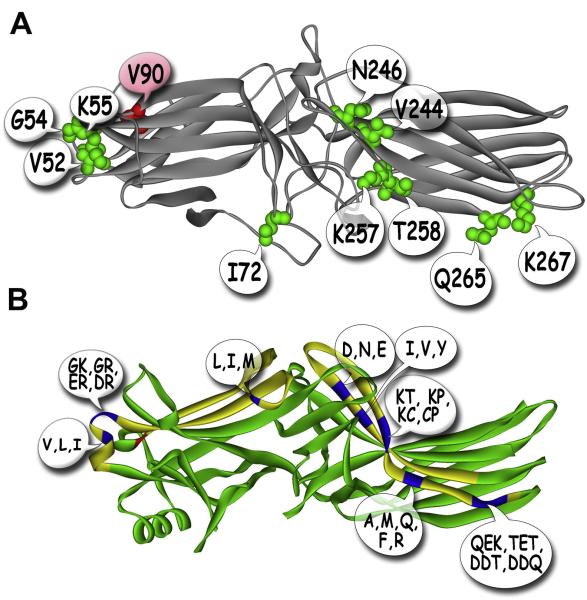
Visual arrestin-1 plays a key role in the rapid and reproducible shutoff of rhodopsin signaling. Its highly selective binding to light-activated phosphorylated rhodopsin is an integral part of the functional perfection of rod photoreceptors. Structure-function studies revealed key elements of the sophisticated molecular mechanism ensuring arrestin-1 selectivity and paved the way to the targeted manipulation of the arrestin-1 molecule to design mutants that can compensate for congenital defects in rhodopsin phosphorylation. Arrestin-1 self-association and light-dependent translocation in photoreceptor cells work together to keep a constant supply of active rhodopsin-binding arrestin-1 monomer in the outer segment. Recent discoveries of arrestin-1 interaction with other signaling proteins suggest that it is a much more versatile signaling regulator than previously thought, affecting the function of the synaptic terminals and rod survival. Elucidation of the fine molecular mechanisms of arrestin-1 interactions with rhodopsin and other binding partners is necessary for the comprehensive understanding of rod function and for devising novel molecular tools and therapeutic approaches to the treatment of visual disorders.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
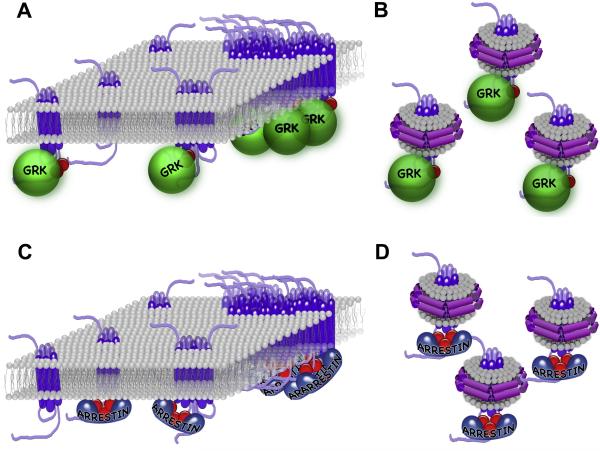
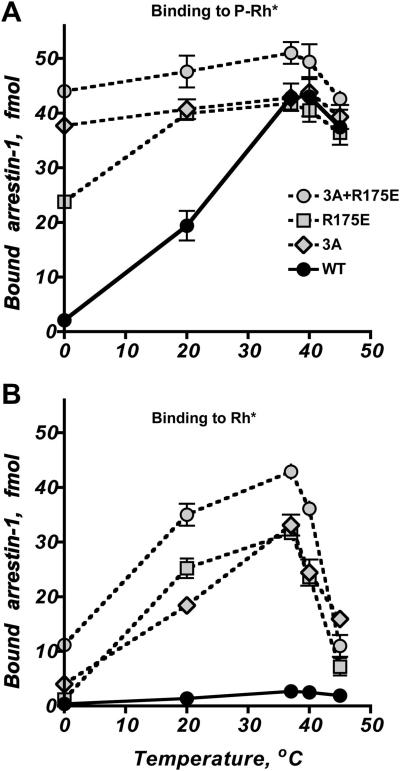
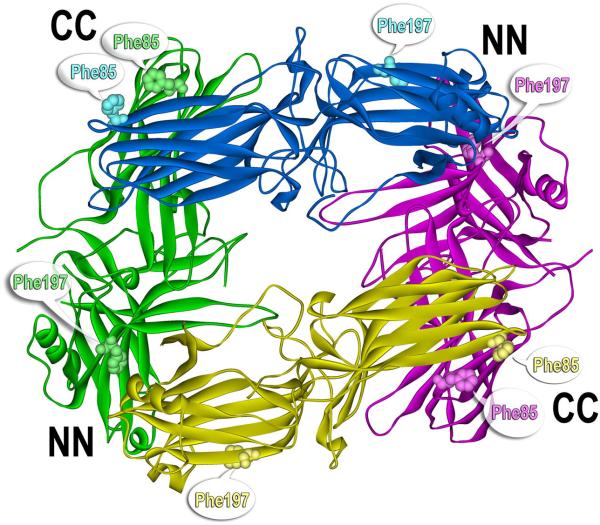
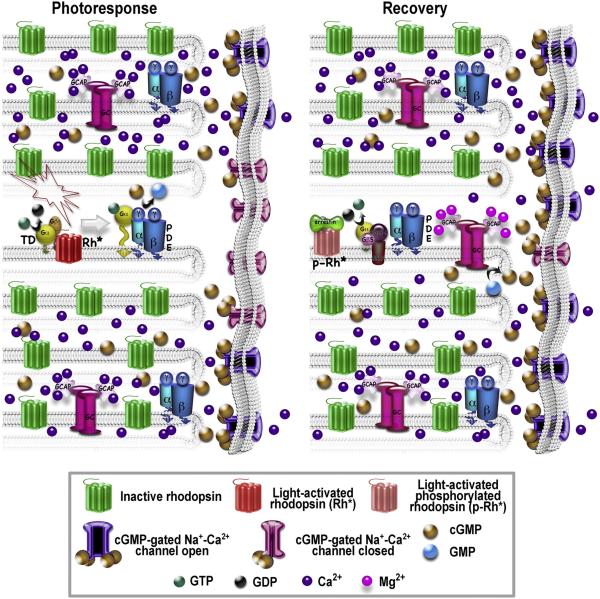
References
-
- Banerjee S, Huber T, Sakmar TP. Rapid incorporation of functional rhodopsin into nanoscale apolipoprotein bound bilayer (NABB) particles. J Mol Biol. 2008;377:1067–1081. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
