

An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance
- PMID: 21828272
- PMCID: PMC3182026
- DOI: 10.1101/gad.17269211
An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance
Erratum in
- Genes Dev. 2011 Sep 15;25(18):1997. Fellman, Christof [corrected to Fellmann, Christof]
Abstract
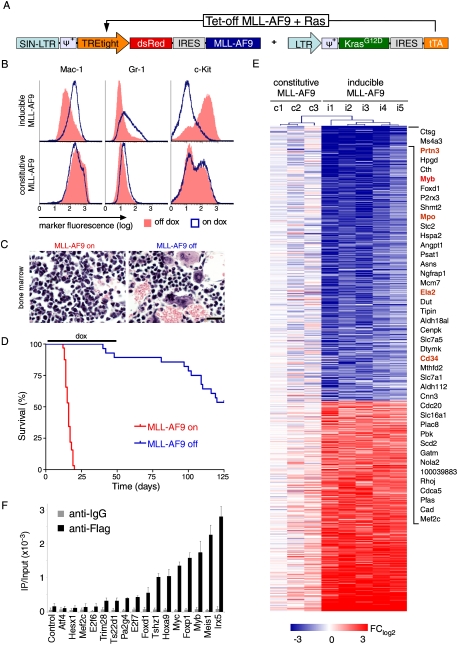
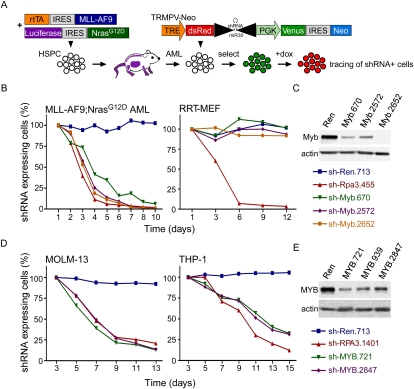
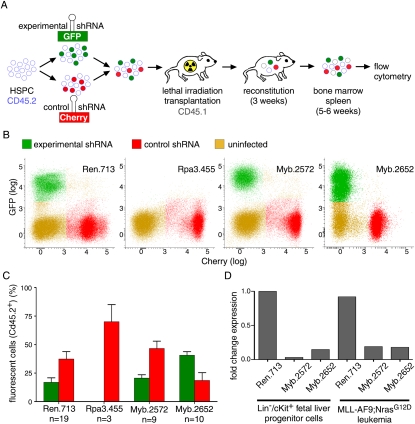
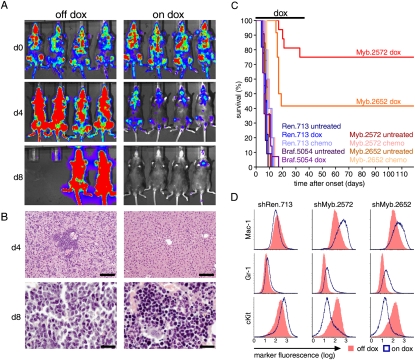
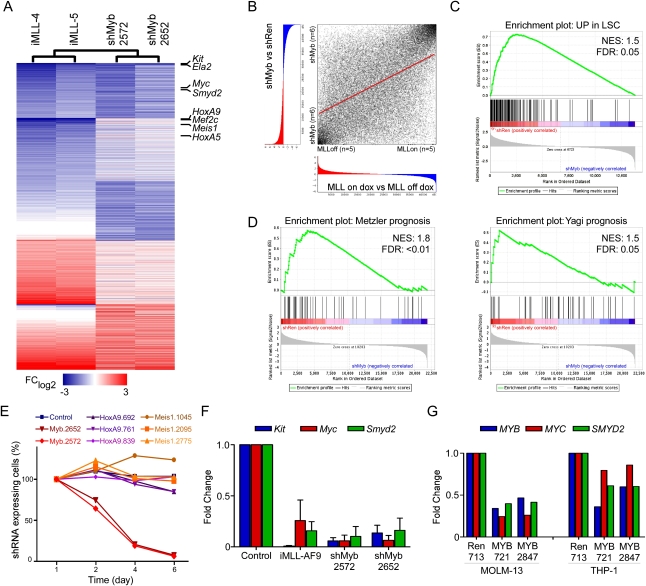
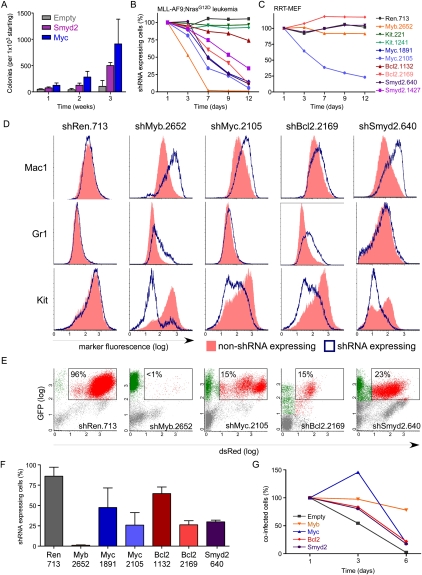
Although human cancers have complex genotypes and are genomically unstable, they often remain dependent on the continued presence of single-driver mutations-a phenomenon dubbed "oncogene addiction." Such dependencies have been demonstrated in mouse models, where conditional expression systems have revealed that oncogenes able to initiate cancer are often required for tumor maintenance and progression, thus validating the pathways they control as therapeutic targets. Here, we implement an integrative approach that combines genetically defined mouse models, transcriptional profiling, and a novel inducible RNAi platform to characterize cellular programs that underlie addiction to MLL-AF9-a fusion oncoprotein involved in aggressive forms of acute myeloid leukemia (AML). We show that MLL-AF9 contributes to leukemia maintenance by enforcing a Myb-coordinated program of aberrant self-renewal involving genes linked to leukemia stem cell potential and poor prognosis in human AML. Accordingly, partial and transient Myb suppression precisely phenocopies MLL-AF9 withdrawal and eradicates aggressive AML in vivo without preventing normal myelopoiesis, indicating that strategies to inhibit Myb-dependent aberrant self-renewal programs hold promise as effective and cancer-specific therapeutics. Together, our results identify Myb as a critical mediator of oncogene addiction in AML, delineate relevant Myb target genes that are amenable to pharmacologic inhibition, and establish a general approach for dissecting oncogene addiction in vivo.
Figures
References
-
- Bender TP, Kremer CS, Kraus M, Buch T, Rajewsky K 2004. Critical functions for c-Myb at three checkpoints during thymocyte development. Nat Immunol 5: 721–729 - PubMed
-
- Chandra P, Luthra R, Zuo Z, Yao H, Ravandi F, Reddy N, Garcia-Manero G, Kantarjian H, Jones D 2010. Acute myeloid leukemia with t(9;11)(p21-22;q23): common properties of dysregulated ras pathway signaling and genomic progression characterize de novo and therapy-related cases. Am J Clin Pathol 133: 686–693 - PubMed
-
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O'Hagan R, Pantginis J, Zhou H, et al. 1999. Essential role for oncogenic Ras in tumour maintenance. Nature 400: 468–472 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials