

LGI2 truncation causes a remitting focal epilepsy in dogs
- PMID: 21829378
- PMCID: PMC3145619
- DOI: 10.1371/journal.pgen.1002194
LGI2 truncation causes a remitting focal epilepsy in dogs
Abstract
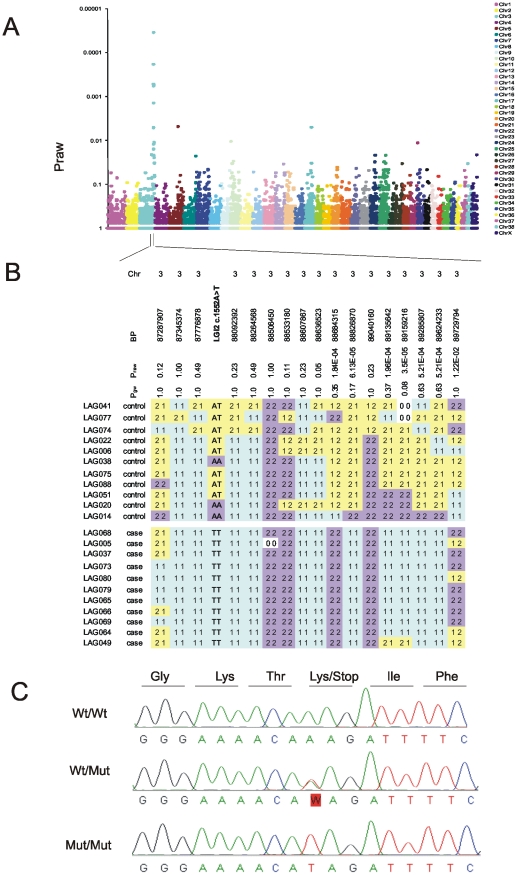
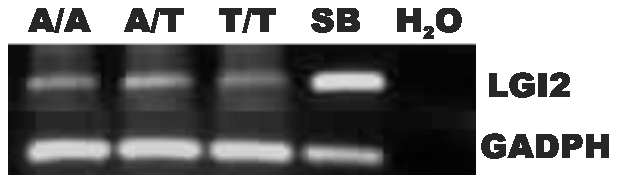
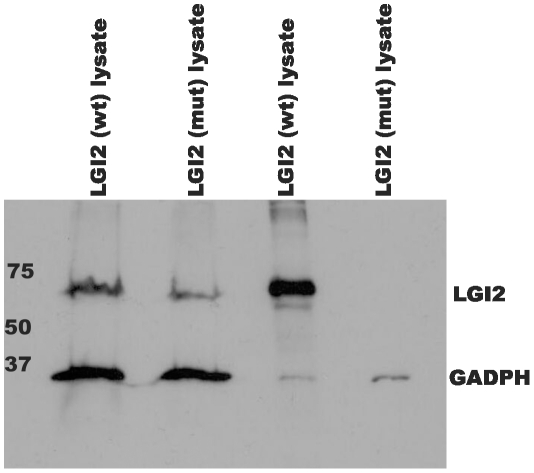
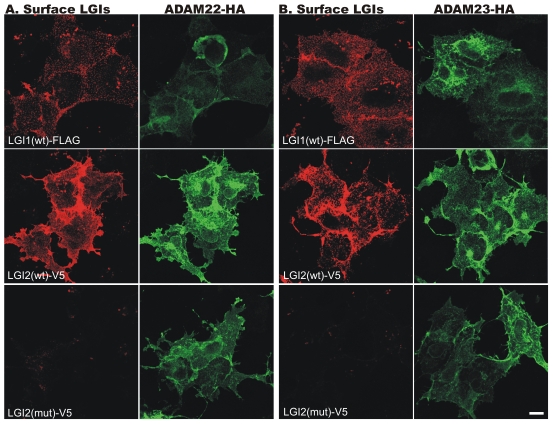
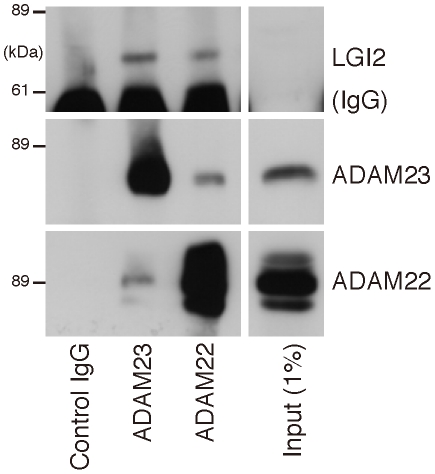
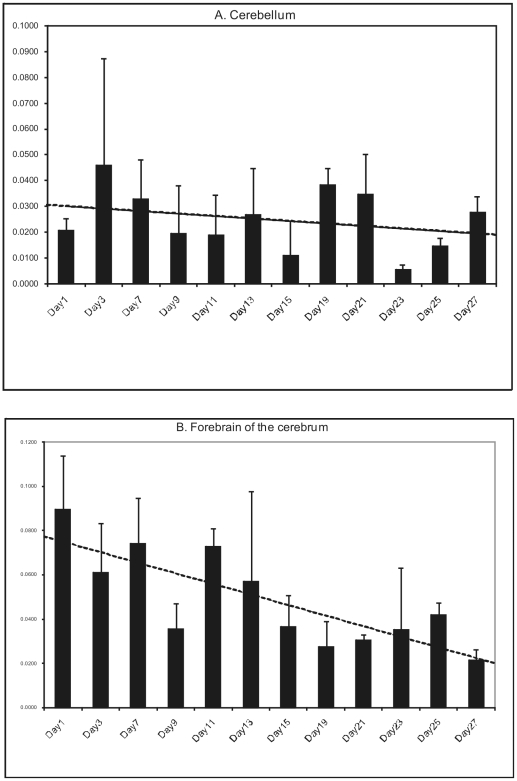
One quadrillion synapses are laid in the first two years of postnatal construction of the human brain, which are then pruned until age 10 to 500 trillion synapses composing the final network. Genetic epilepsies are the most common neurological diseases with onset during pruning, affecting 0.5% of 2-10-year-old children, and these epilepsies are often characterized by spontaneous remission. We previously described a remitting epilepsy in the Lagotto romagnolo canine breed. Here, we identify the gene defect and affected neurochemical pathway. We reconstructed a large Lagotto pedigree of around 34 affected animals. Using genome-wide association in 11 discordant sib-pairs from this pedigree, we mapped the disease locus to a 1.7 Mb region of homozygosity in chromosome 3 where we identified a protein-truncating mutation in the Lgi2 gene, a homologue of the human epilepsy gene LGI1. We show that LGI2, like LGI1, is neuronally secreted and acts on metalloproteinase-lacking members of the ADAM family of neuronal receptors, which function in synapse remodeling, and that LGI2 truncation, like LGI1 truncations, prevents secretion and ADAM interaction. The resulting epilepsy onsets at around seven weeks (equivalent to human two years), and remits by four months (human eight years), versus onset after age eight in the majority of human patients with LGI1 mutations. Finally, we show that Lgi2 is expressed highly in the immediate post-natal period until halfway through pruning, unlike Lgi1, which is expressed in the latter part of pruning and beyond. LGI2 acts at least in part through the same ADAM receptors as LGI1, but earlier, ensuring electrical stability (absence of epilepsy) during pruning years, preceding this same function performed by LGI1 in later years. LGI2 should be considered a candidate gene for common remitting childhood epilepsies, and LGI2-to-LGI1 transition for mechanisms of childhood epilepsy remission.
Conflict of interest statement
A DNA test for the Lagotto breed is commercially available through Genoscoper Oy (Ltd), which is partly owned by HL.
Figures
References
-
- Huttenlocher PR. Morphometric study of human cerebral cortex development. Neuropsychologia. 1990;28(6):517–527. - PubMed
-
- Watson RE, Desesso JM, Hurtt ME, Cappon GD. Postnatal growth and morphological development of the brain: A species comparison. Birth Defects Res B Dev Reprod Toxicol. 2006;77(5):471–484. - PubMed
-
- Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, et al., editors. John Libbey Eurotext; 2005. Epileptic syndromes in infancy, childhood and adolescence (4th edition).616
-
- Turnbull J, Lohi H, Kearney JA, Rouleau GA, Delgado-Escueta AV, et al. Sacred disease secrets revealed: The genetics of human epilepsy. Hum Mol Genet 14 Spec No. 2005;2:2491–2500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
