

Differential gene expression in the siphonophore Nanomia bijuga (Cnidaria) assessed with multiple next-generation sequencing workflows
- PMID: 21829563
- PMCID: PMC3146525
- DOI: 10.1371/journal.pone.0022953
Differential gene expression in the siphonophore Nanomia bijuga (Cnidaria) assessed with multiple next-generation sequencing workflows
Abstract
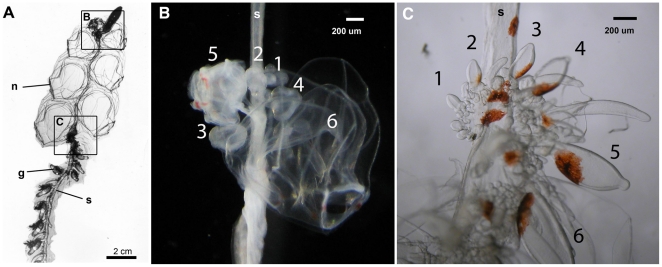
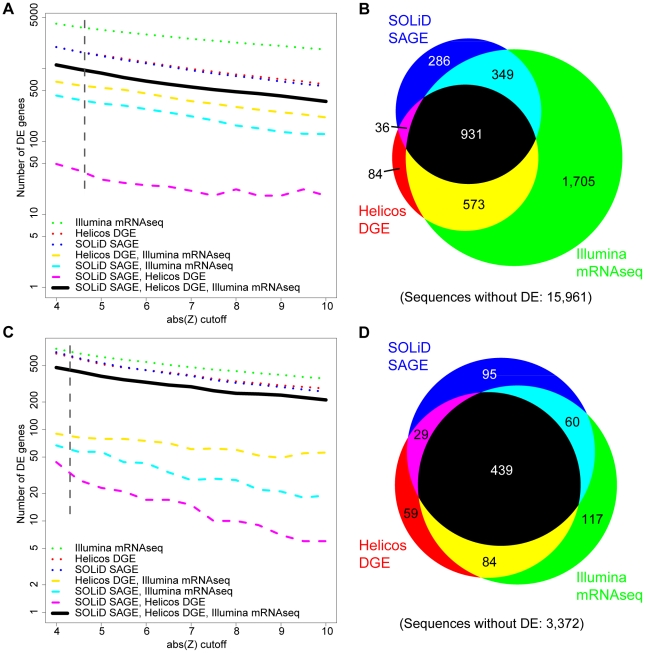
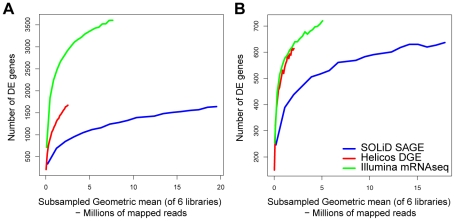
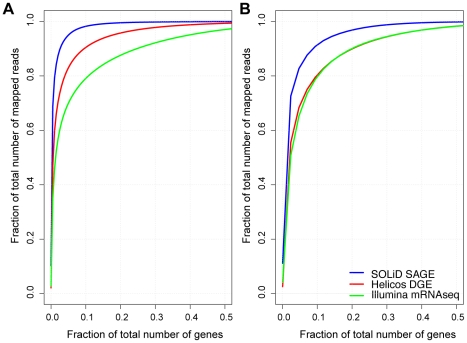
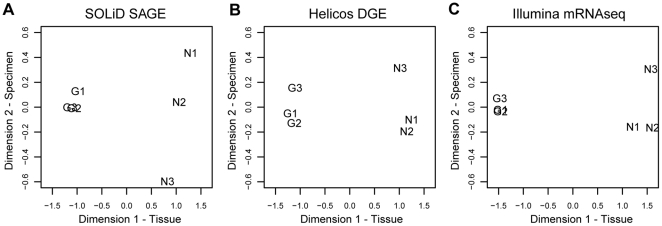
We investigated differential gene expression between functionally specialized feeding polyps and swimming medusae in the siphonophore Nanomia bijuga (Cnidaria) with a hybrid long-read/short-read sequencing strategy. We assembled a set of partial gene reference sequences from long-read data (Roche 454), and generated short-read sequences from replicated tissue samples that were mapped to the references to quantify expression. We collected and compared expression data with three short-read expression workflows that differ in sample preparation, sequencing technology, and mapping tools. These workflows were Illumina mRNA-Seq, which generates sequence reads from random locations along each transcript, and two tag-based approaches, SOLiD SAGE and Helicos DGE, which generate reads from particular tag sites. Differences in expression results across workflows were mostly due to the differential impact of missing data in the partial reference sequences. When all 454-derived gene reference sequences were considered, Illumina mRNA-Seq detected more than twice as many differentially expressed (DE) reference sequences as the tag-based workflows. This discrepancy was largely due to missing tag sites in the partial reference that led to false negatives in the tag-based workflows. When only the subset of reference sequences that unambiguously have tag sites was considered, we found broad congruence across workflows, and they all identified a similar set of DE sequences. Our results are promising in several regards for gene expression studies in non-model organisms. First, we demonstrate that a hybrid long-read/short-read sequencing strategy is an effective way to collect gene expression data when an annotated genome sequence is not available. Second, our replicated sampling indicates that expression profiles are highly consistent across field-collected animals in this case. Third, the impacts of partial reference sequences on the ability to detect DE can be mitigated through workflow choice and deeper reference sequencing.
Conflict of interest statement
Figures
References
-
- Mackie GO, editor. The Lower Metazoa: University of California Press; 1963. pp. 329–337.
-
- Mackie GO, Pugh PR, Purcell JE. Siphonophore Biology. Adv Mar Biol. 1987;24:97–262.
-
- Dunn C. Siphonophores. Current Biology. 2009;19:R233–R234. - PubMed
-
- Dunn CW, Wagner GP. The evolution of colony-level development in the Siphonophora (Cnidaria : Hydrozoa). Development Genes and Evolution. 2006;216:743–754. - PubMed
-
- Harbers M, Carninci P. Tag-based approaches for transcriptome research and genome annotation. Nature Methods. 2005;2:495–502. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
