

Copy number variation in familial Parkinson disease
- PMID: 21829596
- PMCID: PMC3149037
- DOI: 10.1371/journal.pone.0020988
Copy number variation in familial Parkinson disease
Abstract
Copy number variants (CNVs) are known to cause Mendelian forms of Parkinson disease (PD), most notably in SNCA and PARK2. PARK2 has a recessive mode of inheritance; however, recent evidence demonstrates that a single CNV in PARK2 (but not a single missense mutation) may increase risk for PD. We recently performed a genome-wide association study for PD that excluded individuals known to have either a LRRK2 mutation or two PARK2 mutations. Data from the Illumina370Duo arrays were re-clustered using only white individuals with high quality intensity data, and CNV calls were made using two algorithms, PennCNV and QuantiSNP. After quality assessment, the final sample included 816 cases and 856 controls. Results varied between the two CNV calling algorithms for many regions, including the PARK2 locus (genome-wide p = 0.04 for PennCNV and p = 0.13 for QuantiSNP). However, there was consistent evidence with both algorithms for two novel genes, USP32 and DOCK5 (empirical, genome-wide p-values<0.001). PARK2 CNVs tended to be larger, and all instances that were molecularly tested were validated. In contrast, the CNVs in both novel loci were smaller and failed to replicate using real-time PCR, MLPA, and gel electrophoresis. The DOCK5 variation is more akin to a VNTR than a typical CNV and the association is likely caused by artifact due to DNA source. DNA for all the cases was derived from whole blood, while the DNA for all controls was derived from lymphoblast cell lines. The USP32 locus contains many SNPs with low minor allele frequency leading to a loss of heterozygosity that may have been spuriously interpreted by the CNV calling algorithms as support for a deletion. Thus, only the CNVs within the PARK2 locus could be molecularly validated and associated with PD susceptibility.
Conflict of interest statement
Figures
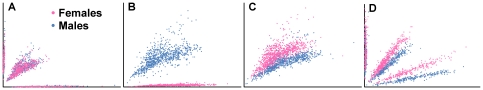
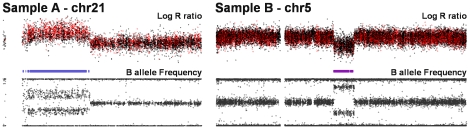
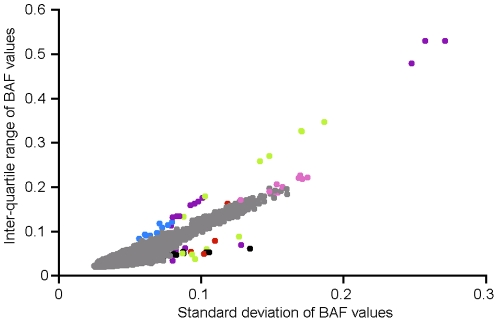
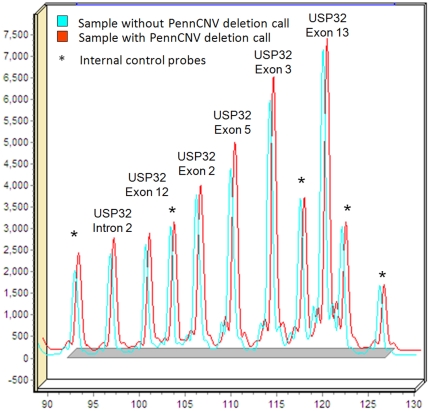

References
-
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. - PubMed
-
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
