

Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure
- PMID: 21831279
- PMCID: PMC3170181
- DOI: 10.1186/1750-1172-6-55
Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure
Abstract
Background: Mucopolysaccharidosis type I (MPS I) is a lysosomal storage disorder that results in the accumulation of glycosaminoglycans causing progressive multi-organ dysfunction. Its clinical spectrum is very broad and varies from the severe Hurler phenotype (MPS I-H) which is characterized by early and progressive central nervous system (CNS) involvement to the attenuated Scheie phenotype (MPS I-S) with no CNS involvement. Indication, optimal timing, safety and efficacy of the two available treatment options for MPS I, enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT), are subject to continuing debate. A European consensus procedure was organized to reach consensus about the use of these two treatment strategies.
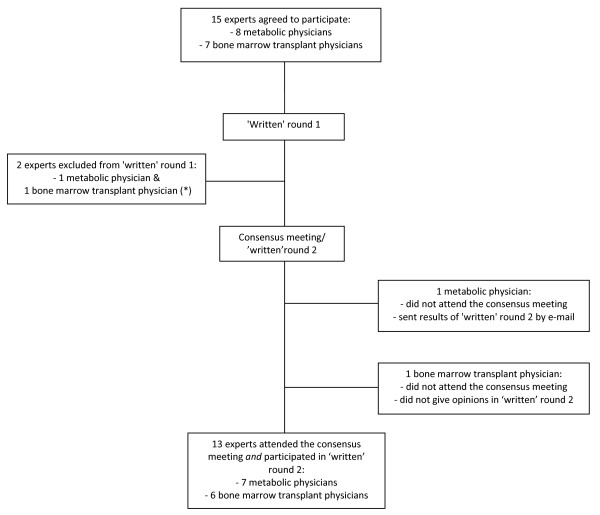
Methods: A panel of specialists, including 8 specialists for metabolic disorders and 7 bone marrow transplant physicians, all with acknowledged expertise in MPS I, participated in a modified Delphi process to develop consensus-based statements on MPS I treatment. Fifteen MPS I case histories were used to initiate the discussion and to anchor decisions around either treatment mode. Before and at the meeting all experts gave their opinion on the cases (YES/NO transplantation) and reasons for their decisions were collected. A set of draft statements on MPS I treatment options composed by a planning committee were discussed and revised during the meeting until full consensus.
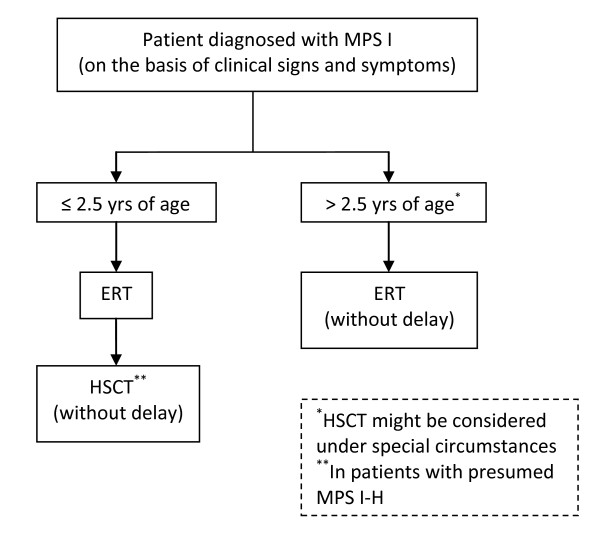
Results: Full consensus was reached on several important issues, including the following: 1) The preferred treatment for patients with MPS I-H diagnosed before age 2.5 yrs is HSCT; 2) In individual patients with an intermediate phenotype HSCT may be considered if there is a suitable donor. However, there are no data on efficacy of HSCT in patients with this phenotype; 3) All MPS I patients including those who have not been transplanted or whose graft has failed may benefit significantly from ERT; 4) ERT should be started at diagnosis and may be of value in patients awaiting HSCT.
Conclusions: This multidisciplinary consensus procedure yielded consensus on the main issues related to therapeutic choices and research for MPS I. This is an important step towards an international, collaborative approach, the only way to obtain useful evidence in rare diseases.
Figures
References
-
- Neufeld EF, Muenzer J. In: The Online Metabolic and Molecular Bases of Inherited Disease. Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, editor. chapter 136. New York: McGraw-Hill; 2007. The mucopolysaccharidoses.
-
- Pastores GM, Arn P, Beck M, Clarke JT, Guffon N, Kaplan P, Muenzer J, Norato DY, Shapiro E, Thomas J, Viskochil D, Wraith JE. The MPS I registry: Design, methodology, and early findings of a global disease registry for monitoring patients with Mucopolysaccharidosis Type I. Mol Genet Metab. 2007;91:37–47. doi: 10.1016/j.ymgme.2007.01.011. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
