

Curaxins: anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT
- PMID: 21832239
- PMCID: PMC6281439
- DOI: 10.1126/scitranslmed.3002530
Curaxins: anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT
Abstract
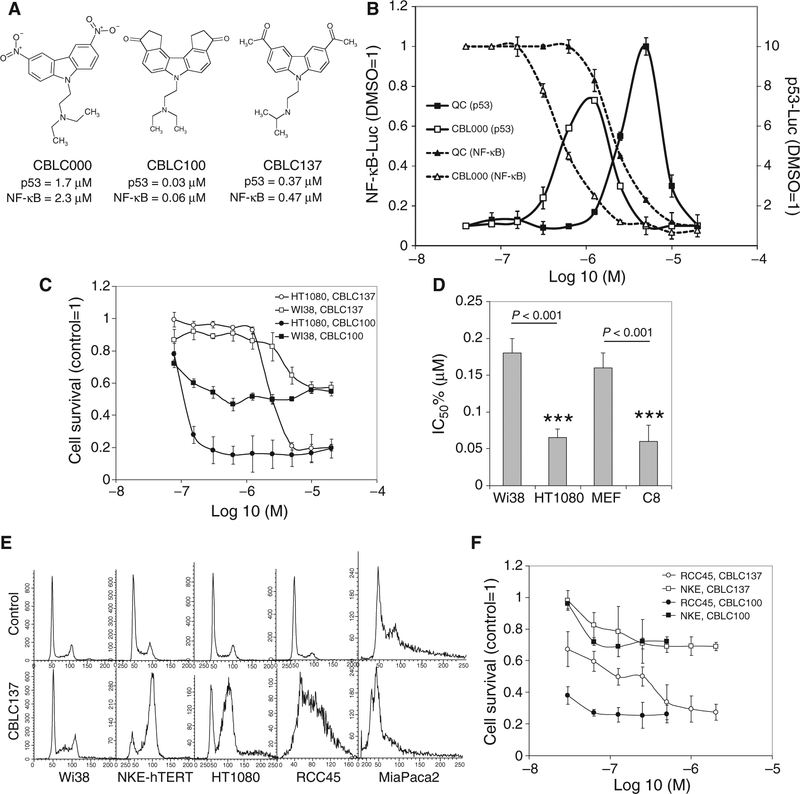
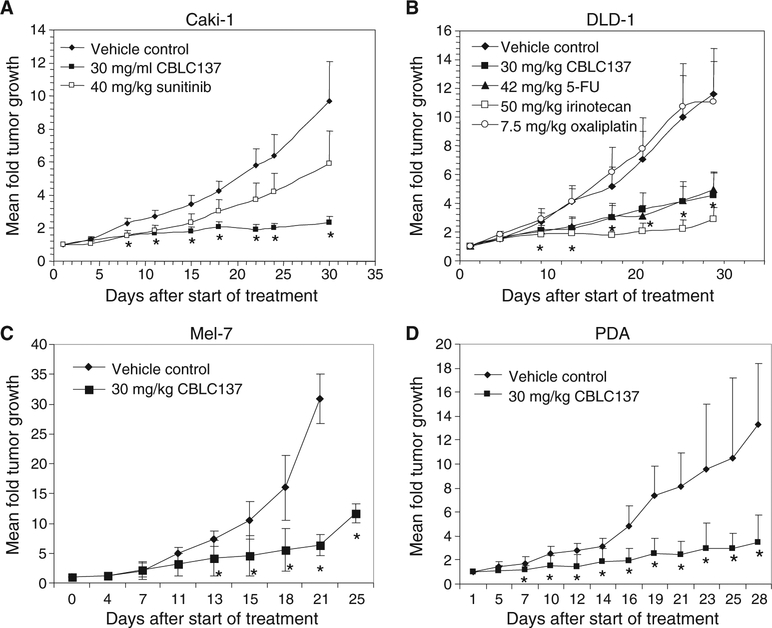
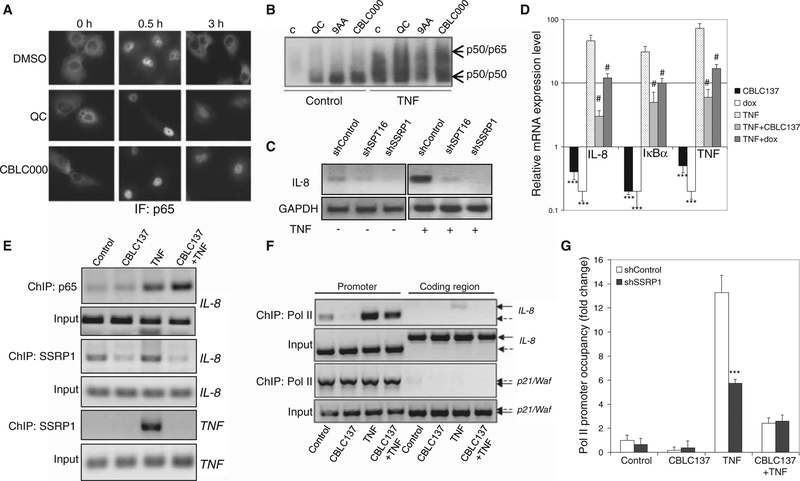
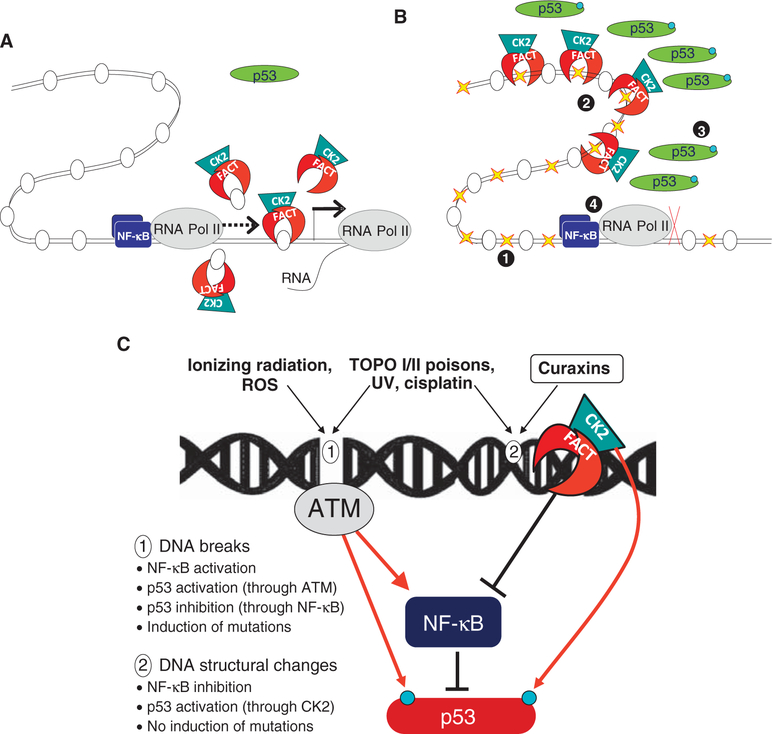
Effective eradication of cancer requires treatment directed against multiple targets. The p53 and nuclear factor κB (NF-κB) pathways are dysregulated in nearly all tumors, making them attractive targets for therapeutic activation and inhibition, respectively. We have isolated and structurally optimized small molecules, curaxins, that simultaneously activate p53 and inhibit NF-κB without causing detectable genotoxicity. Curaxins demonstrated anticancer activity against all tested human tumor xenografts grown in mice. We report here that the effects of curaxins on p53 and NF-κB, as well as their toxicity to cancer cells, result from "chromatin trapping" of the FACT (facilitates chromatin transcription) complex. This FACT inaccessibility leads to phosphorylation of the p53 Ser(392) by casein kinase 2 and inhibition of NF-κB-dependent transcription, which requires FACT activity at the elongation stage. These results identify FACT as a prospective anticancer target enabling simultaneous modulation of several pathways frequently dysregulated in cancer without induction of DNA damage. Curaxins have the potential to be developed into effective and safe anticancer drugs.
Conflict of interest statement
Figures




Comment in
-
Cancer drug discovery faces the FACT.Sci Transl Med. 2011 Aug 10;3(95):95ps34. doi: 10.1126/scitranslmed.3002822. Sci Transl Med. 2011. PMID: 21832237
References
-
- Kawanishi S, Hiraku Y, Amplification of anticancer drug-induced DNA damage and apoptosis by DNA-binding compounds. Curr. Med. Chem. Anticancer Agents 4, 415–419 (2004). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
