

Review
doi: 10.1038/nature10275.
A continuum model for tumour suppression
Affiliations
- PMID: 21833082
- PMCID: PMC3206311
- DOI: 10.1038/nature10275
Item in Clipboard
Review
A continuum model for tumour suppression
Nature.
.
Abstract
This year, 2011, marks the forty-year anniversary of the statistical analysis of retinoblastoma that provided the first evidence that tumorigenesis can be initiated by as few as two mutations. This work provided the foundation for the two-hit hypothesis that explained the role of recessive tumour suppressor genes (TSGs) in dominantly inherited cancer susceptibility syndromes. However, four decades later, it is now known that even partial inactivation of tumour suppressors can critically contribute to tumorigenesis. Here we analyse this evidence and propose a continuum model of TSG function to explain the full range of TSG mutations found in cancer.
Figures
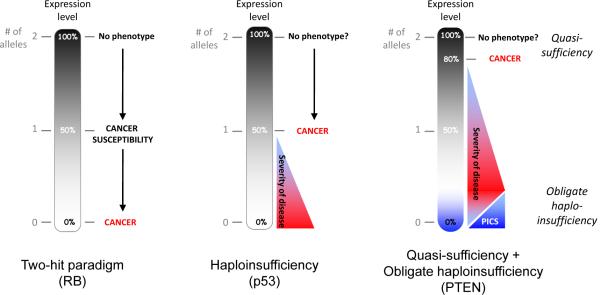
A black gradient represents a continuum of expression that is also related to the number of alleles present (gray numbering). Left, the two-hit paradigm as exemplified by the tumour suppressor, RB. Loss of one allele induces cancer susceptibility; loss of two alleles induces cancer. Middle, classic haploinsufficiency. Loss of one allele is sufficient for induction of cancer. Right, Quasi-sufficiency and obligate haploinsufficiency. Quasi-sufficiency refers to the phenomenon whereby tumour suppression is impaired after subtle expression downregulation without loss of even one allele. Obligate haploinsufficiency occurs when TSG haploinsufficiency is more tumourigenic than complete loss of the TSG, usually due to the activation of fail-safe mechanisms following complete loss of TSG expression.
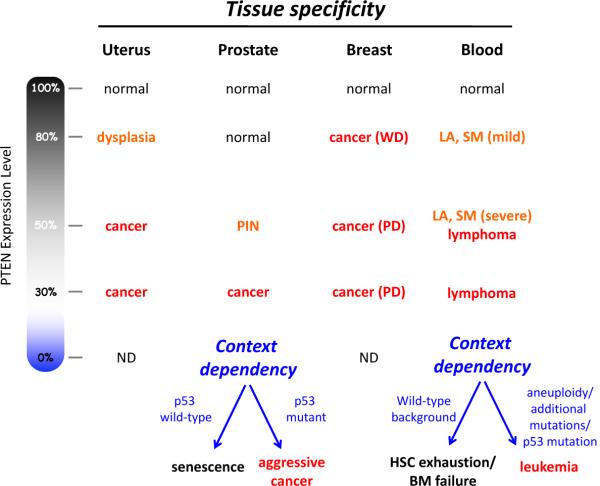
The phenotypic outcome of a reduction in PTEN expression is differentially manifested depending upon tissue type and genetic background. WD, well-differentiated. PD, poorly-differentiated. LA, lymphadenopathy. SM, splenomegaly. HSC, hematopoietic stem cell. BM, bone marrow. The effect of complete loss of PTEN is highly context dependent due to the obligate haploinsufficiency caused by PTEN loss-induced cellular senescence (PICS). Data summarized here come from multiple groups and studies of genetically-engineered mice with differing PTEN alleles and expression–,,–.
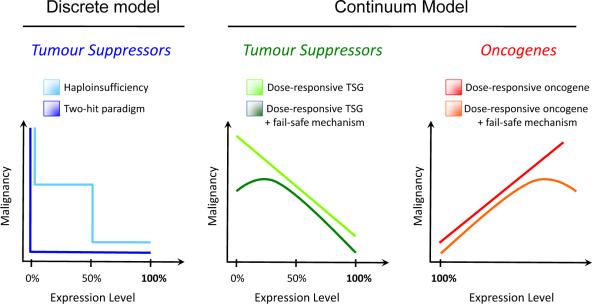
The classical discrete, stepwise model of tumour suppression (left) is contrasted with a continuum model of tumour suppression and oncogenesis (centre and right, respectively). In the discrete model, tumourigenesis is induced by either complete loss of a TSG (two-hit paradigm, dark blue) or after single-copy loss of a TSG (haploinsufficiency, light blue). In contrast, we propose a continuum model (centre and right), in which tumour suppression is related to a continuum of TSG expression, rather than to discrete changes in DNA copy number. A continuum of increasing TSG expression will generally be negatively correlated with malignancy (centre, light green) whereas increasing oncogene expression will generally be positively correlated with malignancy (right, red). A linear relationship is depicted for schematic purposes, but the dose-response relationship need not be linear. In some cases, fail-safe mechanisms are induced by complete loss of TSG expression or by massive oncogene overexpression. In these cases, complete loss of TSG expression (centre, dark green) or massive overexpression of an oncogene (right, orange) will be negatively correlated with malignancy, as shown.
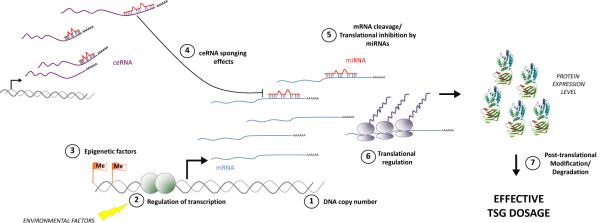
The interaction between coding and non-coding factors determines final TSG dosage. Classic mechanisms such as DNA copy number (1), transcriptional regulation (2), and epigenetic silencing can impact expression of TSG mRNA. TSG mRNA level or translation into protein is then regulated by miRNAs (4). The availability of miRNAs for TSG downregulation is further regulated by ceRNA-mediated sponging effects (3). Finally, additional translational regulation (6) or post-translational modifications contribute to the final protein expression, function and effective dosage. The protein structure shown is PTEN (PDB ID: 1D5R).
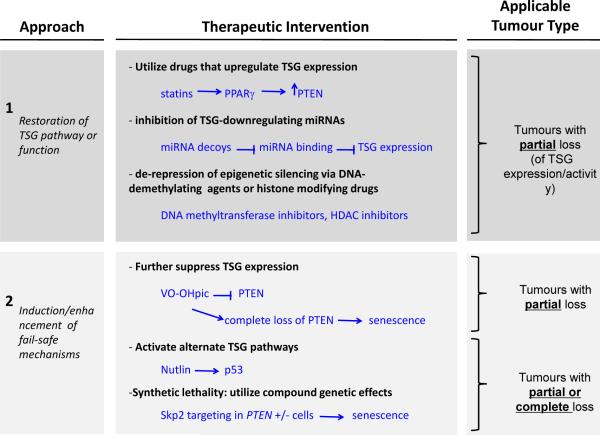
Tumour genotype ultimately determines which therapies can be considered for intervention. Broadly, two general approaches can be taken: restoration of the TSG pathway or function (upper panel) or induction of fail-safe mechanisms by complete downregulation of the TSG (lower panel). In tumours with downregulation of TSG expression in the absence of complete mutation or deletion, expression of the TSG can be attempted using drugs that induce the TSG transcription, inhibit miRNA-induced TSG downregulation, or relieve epigenetic silencing. Alternatively, fail-safe mechanisms may be induced by further downregulation of the TSG, such as inhibition of PTEN by the small molecule VO-OHpic. In tumours with mutation or complete loss of the TSG, activation of other TSG pathways could be pursued. Alternatively, if a synthetic lethality relationship with the TSG is known, then the synthetic lethal gene can be targeted to induce cell death in the cells with complete loss of the TSG.
References
-
- Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85–109. - PubMed
-
- Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–3. - PubMed
-
- Heisterkamp N, et al. Localization of the c-ab1 oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature. 1983;306:239–42. - PubMed
-
- Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet. 1977;1:549–50. - PubMed
-
- Pandolfi PP, et al. Structure and origin of the acute promyelocytic leukemia myl/RAR alpha cDNA and characterization of its retinoid-binding and transactivation properties. Oncogene. 1991;6:1285–92. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
