

Organellar dysfunction in the pathogenesis of pancreatitis
- PMID: 21834686
- PMCID: PMC3183656
- DOI: 10.1089/ars.2011.4068
Organellar dysfunction in the pathogenesis of pancreatitis
Abstract
Significance: Acute pancreatitis is an inflammatory disease of exocrine pancreas that carries considerable morbidity and mortality; its pathophysiology remains poorly understood. During the past decade, new insights have been gained into signaling pathways and molecules that mediate the inflammatory response of pancreatitis and death of acinar cells (the main exocrine pancreas cell type). By contrast, much less is known about the acinar cell organellar damage in pancreatitis and how it contributes to the disease pathogenesis.
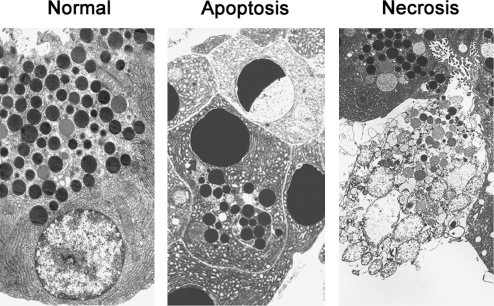
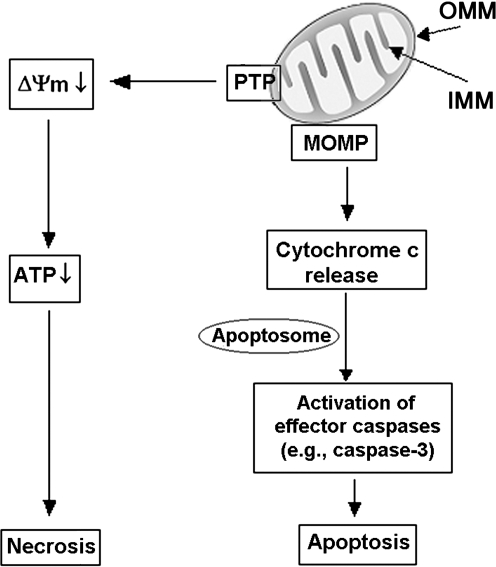
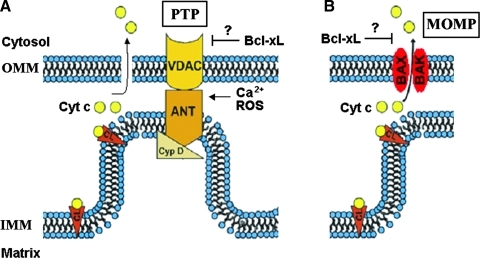
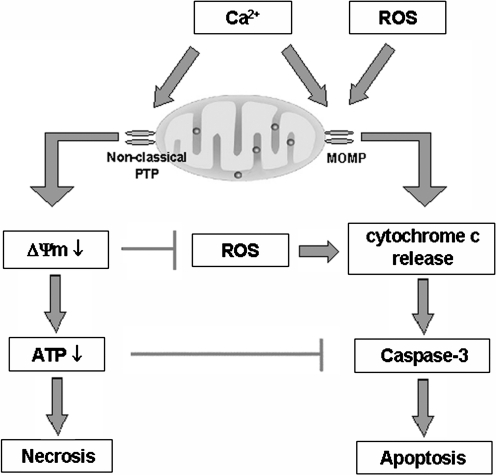
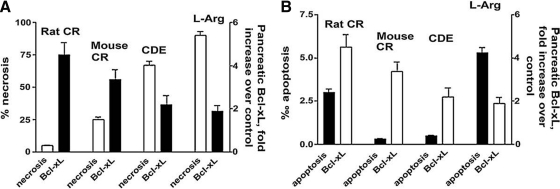
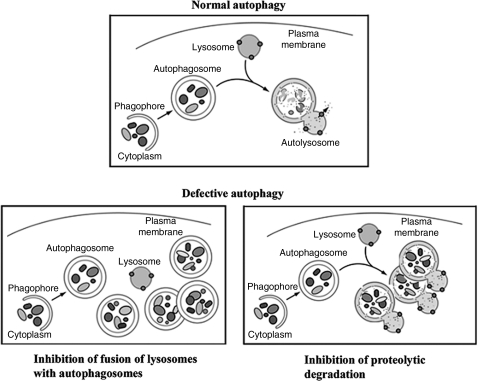
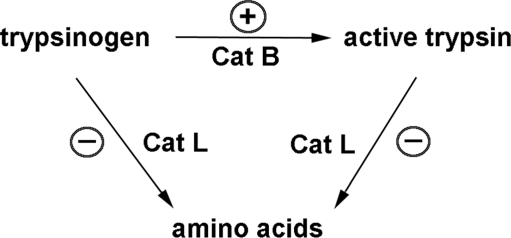
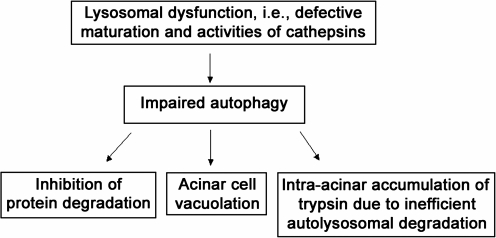
Recent advances: This review summarizes recent findings from our group, obtained on experimental in vivo and ex vivo models, which reveal disordering of key cellular organelles, namely, mitochondria, autophagosomes, and lysosomes, in pancreatitis. Our results indicate a critical role for mitochondrial permeabilization in determining the balance between apoptosis and necrosis in pancreatitis, and thus the disease severity. We further investigate how the mitochondrial dysfunction (and hence acinar cell death) is regulated by Ca(2+), reactive oxygen species, and Bcl-xL, in relation to specific properties of pancreatic mitochondria. Our results also reveal that autophagy, the principal cellular degradative, lysosome-driven pathway, is impaired in pancreatitis due to inefficient lysosomal function, and that impaired autophagy mediates two key pathological responses of pancreatitis-accumulation of vacuoles in acinar cells and the abnormal, intra-acinar activation of digestive enzymes such as trypsinogen.
Critical issues and future directions: The findings discussed in this review indicate critical roles for mitochondrial and autophagic/lysosomal dysfunctions in the pathogenesis of pancreatitis and delineate directions for detailed investigations into the molecular events that underlie acinar cell organellar damage.
Figures
References
-
- Adams JM. Cory S. Apoptosomes: engines for caspase activation. Curr Opin Cell Biol. 2002;14:715–720. - PubMed
-
- Adler G. Rohr G. Kern HF. Alteration of membrane fusion as a cause of acute pancreatitis in the rat. Dig Dis Sci. 1982;27:993–1002. - PubMed
-
- Aho HJ. Nevalainen TJ. Havia VT. Heinonen RJ. Aho AJ. Human acute pancreatitis: a light and electron microscopic study. Acta Pathol Microbiol Immunol Scand A. 1982;90:367–373. - PubMed
-
- Andreyev AY. Kushnareva YE. Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005;70:200–214. - PubMed
-
- Armstrong JS. Mitochondrial membrane permeabilization: the sine qua non for cell death. Bioessays. 2006;28:253–260. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
