

Studying gene and gene-environment effects of uncommon and common variants on continuous traits: a marker-set approach using gene-trait similarity regression
- PMID: 21835306
- PMCID: PMC3155192
- DOI: 10.1016/j.ajhg.2011.07.007
Studying gene and gene-environment effects of uncommon and common variants on continuous traits: a marker-set approach using gene-trait similarity regression
Abstract
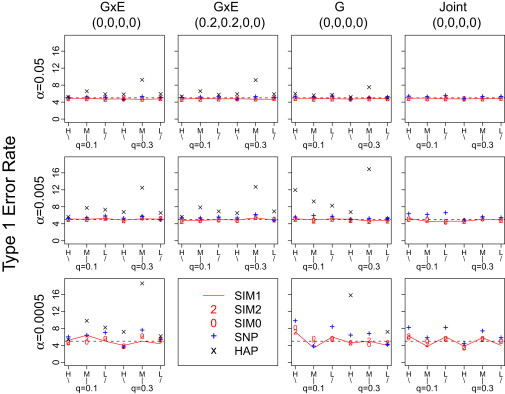
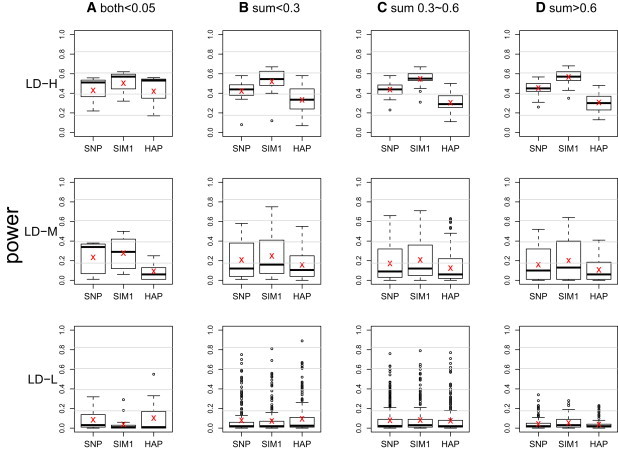
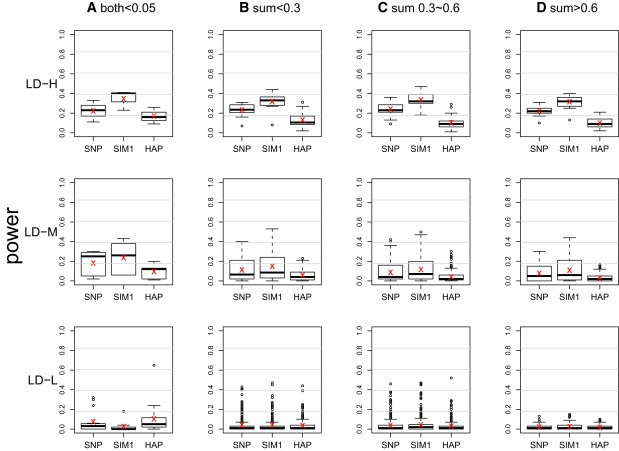
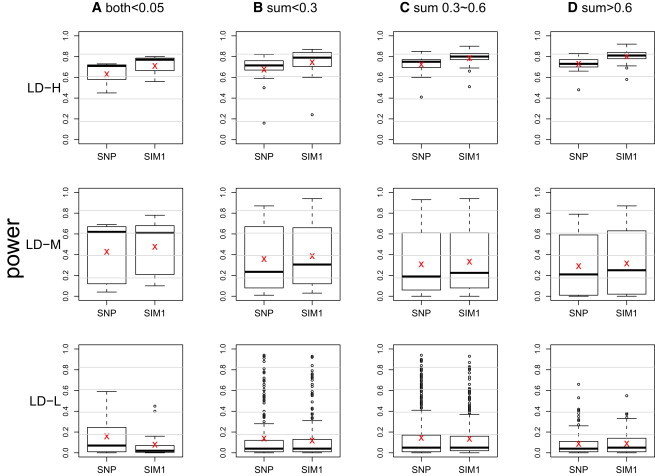
Genomic association analyses of complex traits demand statistical tools that are capable of detecting small effects of common and rare variants and modeling complex interaction effects and yet are computationally feasible. In this work, we introduce a similarity-based regression method for assessing the main genetic and interaction effects of a group of markers on quantitative traits. The method uses genetic similarity to aggregate information from multiple polymorphic sites and integrates adaptive weights that depend on allele frequencies to accomodate common and uncommon variants. Collapsing information at the similarity level instead of the genotype level avoids canceling signals that have the opposite etiological effects and is applicable to any class of genetic variants without the need for dichotomizing the allele types. To assess gene-trait associations, we regress trait similarities for pairs of unrelated individuals on their genetic similarities and assess association by using a score test whose limiting distribution is derived in this work. The proposed regression framework allows for covariates, has the capacity to model both main and interaction effects, can be applied to a mixture of different polymorphism types, and is computationally efficient. These features make it an ideal tool for evaluating associations between phenotype and marker sets defined by linkage disequilibrium (LD) blocks, genes, or pathways in whole-genome analysis.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
Gene-trait similarity regression for multimarker-based association analysis.Biometrics. 2009 Sep;65(3):822-32. doi: 10.1111/j.1541-0420.2008.01176.x. Epub 2009 Feb 4. Biometrics. 2009. PMID: 19210740 Free PMC article.
-
Assessing gene-environment interactions for common and rare variants with binary traits using gene-trait similarity regression.Genetics. 2015 Mar;199(3):695-710. doi: 10.1534/genetics.114.171686. Epub 2015 Jan 12. Genetics. 2015. PMID: 25585620 Free PMC article.
-
Functional linear models for association analysis of quantitative traits.Genet Epidemiol. 2013 Nov;37(7):726-42. doi: 10.1002/gepi.21757. Genet Epidemiol. 2013. PMID: 24130119 Free PMC article.
-
Regression models for linkage: issues of traits, covariates, heterogeneity, and interaction.Hum Hered. 2003;55(2-3):86-96. doi: 10.1159/000072313. Hum Hered. 2003. PMID: 12931047 Review.
-
On selecting markers for association studies: patterns of linkage disequilibrium between two and three diallelic loci.Genet Epidemiol. 2003 Jan;24(1):57-67. doi: 10.1002/gepi.10217. Genet Epidemiol. 2003. PMID: 12508256 Review.
Cited by
-
Powerful Set-Based Gene-Environment Interaction Testing Framework for Complex Diseases.Genet Epidemiol. 2015 Dec;39(8):609-18. doi: 10.1002/gepi.21908. Epub 2015 Jun 10. Genet Epidemiol. 2015. PMID: 26095235 Free PMC article.
-
A unified powerful set-based test for sequencing data analysis of GxE interactions.Biostatistics. 2017 Jan;18(1):119-131. doi: 10.1093/biostatistics/kxw034. Epub 2016 Jul 28. Biostatistics. 2017. PMID: 27474101 Free PMC article.
-
Comparison of statistical tests for association between rare variants and binary traits.PLoS One. 2012;7(8):e42530. doi: 10.1371/journal.pone.0042530. Epub 2012 Aug 9. PLoS One. 2012. PMID: 22912707 Free PMC article.
-
Beyond the fourth wave of genome-wide obesity association studies.Nutr Diabetes. 2012 Jul 30;2(7):e37. doi: 10.1038/nutd.2012.9. Nutr Diabetes. 2012. PMID: 23168490 Free PMC article.
-
Analysis of gene-gene interactions using gene-trait similarity regression.Hum Hered. 2014;78(1):17-26. doi: 10.1159/000360161. Epub 2014 Jun 21. Hum Hered. 2014. PMID: 24969398 Free PMC article.
References
-
- Fisher R.A. Oliver and Boyd; London: 1932. Statistical methods for research workers.
-
- Gauderman W.J., Murcray C., Gilliland F., Conti D.V. Testing association between disease and multiple SNPs in a candidate gene. Genet. Epidemiol. 2007;31:383–395. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA85848/CA/NCI NIH HHS/United States
- R01 MH084022/MH/NIMH NIH HHS/United States
- R01 CA085848/CA/NCI NIH HHS/United States
- R01 ES019876/ES/NIEHS NIH HHS/United States
- 090532/WT_/Wellcome Trust/United Kingdom
- M01 RR007122/RR/NCRR NIH HHS/United States
- P01 CA142538/CA/NCI NIH HHS/United States
- R01 MH074027/MH/NIMH NIH HHS/United States
- U01 HG005160/HG/NHGRI NIH HHS/United States
- R37 AI031789-20/AI/NIAID NIH HHS/United States
- R37 AI031789/AI/NIAID NIH HHS/United States
- M01 RR07122/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
