

ParaDock: a flexible non-specific DNA--rigid protein docking algorithm
- PMID: 21835777
- PMCID: PMC3203577
- DOI: 10.1093/nar/gkr620
ParaDock: a flexible non-specific DNA--rigid protein docking algorithm
Abstract
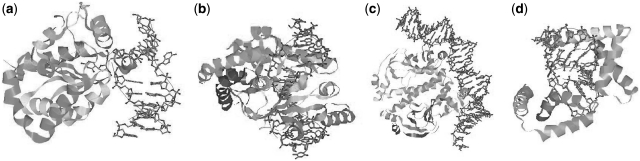
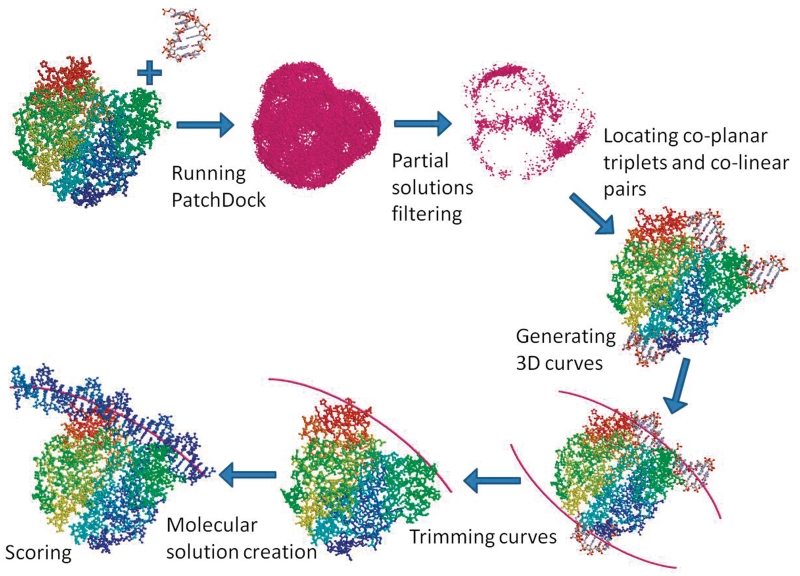
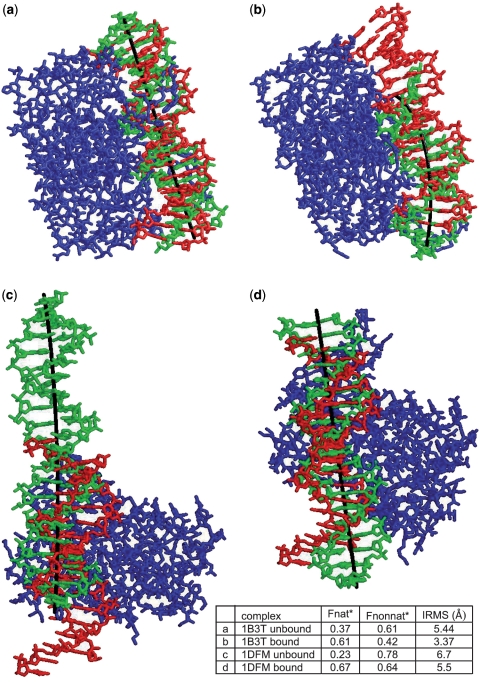
Accurate prediction of protein-DNA complexes could provide an important stepping stone towards a thorough comprehension of vital intracellular processes. Few attempts were made to tackle this issue, focusing on binding patch prediction, protein function classification and distance constraints-based docking. We introduce ParaDock: a novel ab initio protein-DNA docking algorithm. ParaDock combines short DNA fragments, which have been rigidly docked to the protein based on geometric complementarity, to create bent planar DNA molecules of arbitrary sequence. Our algorithm was tested on the bound and unbound targets of a protein-DNA benchmark comprised of 47 complexes. With neither addressing protein flexibility, nor applying any refinement procedure, CAPRI acceptable solutions were obtained among the 10 top ranked hypotheses in 83% of the bound complexes, and 70% of the unbound. Without requiring prior knowledge of DNA length and sequence, and within <2 h per target on a standard 2.0 GHz single processor CPU, ParaDock offers a fast ab initio docking solution.
Figures
References
-
- Ohlendorf DH, Matthew JB. Electrostatics and flexibility in protein–DNA interactions. Advan. Biophys. 1985;20:137–151. - PubMed
-
- Nadassy K, Wodak SJ, Janin J. Structural features of protein−nucleic acid recognition sites. Biochemistry. 1999;38:1999–2017. - PubMed
-
- Mandel-Gutfreund Y, Margalit H, Jernigan RL, Zhurkin VB. A role for CH … O interactions in protein–DNA recognition. J. Mol. Biol. 1998;277:1140–1129. - PubMed
