

APPL1 mediates adiponectin-induced LKB1 cytosolic localization through the PP2A-PKCzeta signaling pathway
- PMID: 21835890
- PMCID: PMC3182423
- DOI: 10.1210/me.2011-0082
APPL1 mediates adiponectin-induced LKB1 cytosolic localization through the PP2A-PKCzeta signaling pathway
Abstract
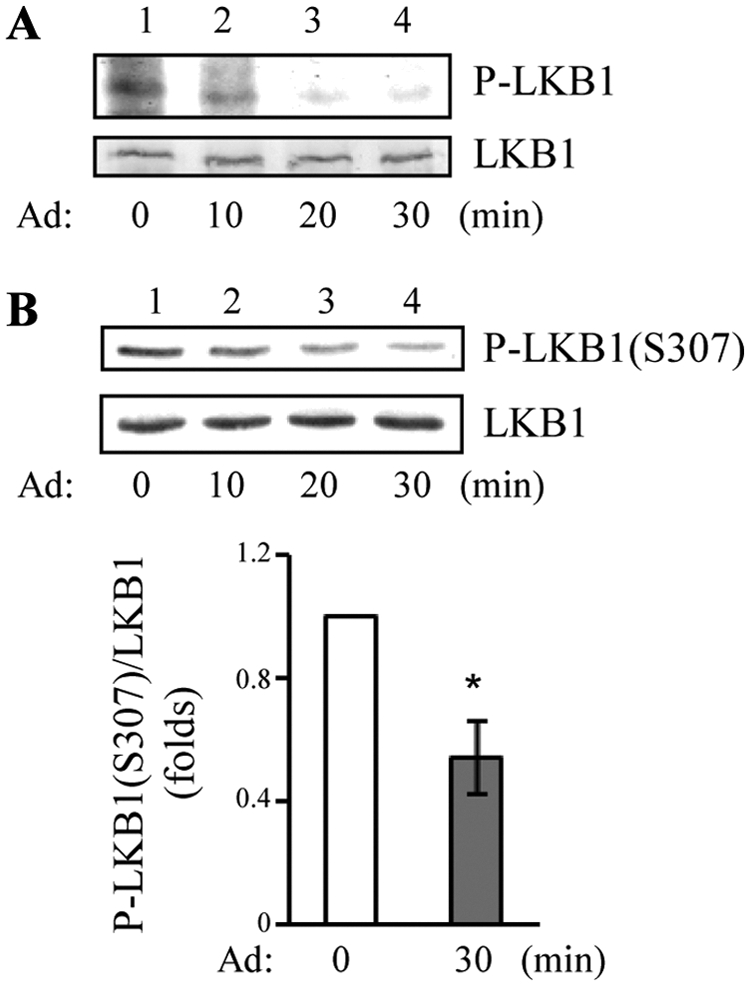
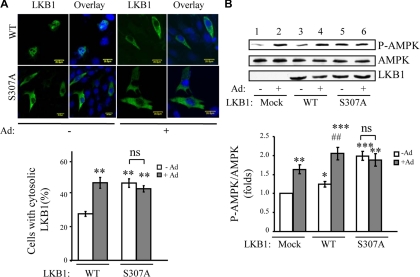
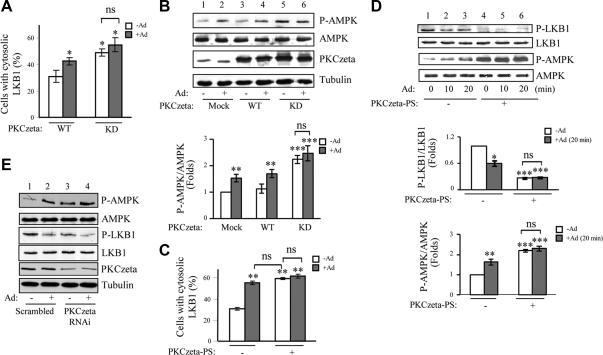
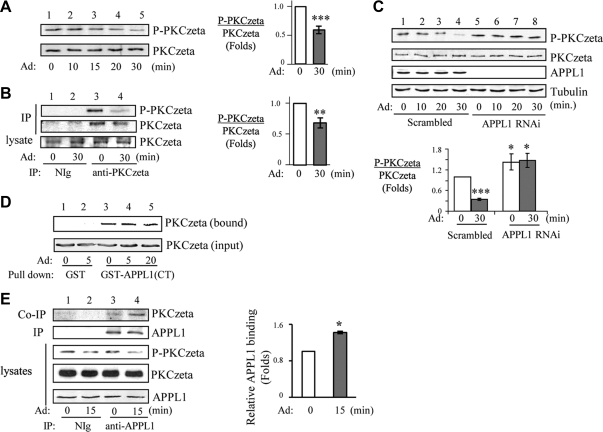
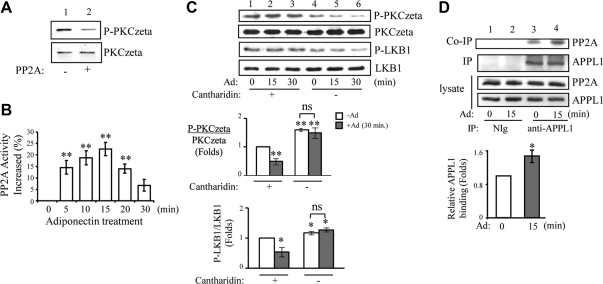
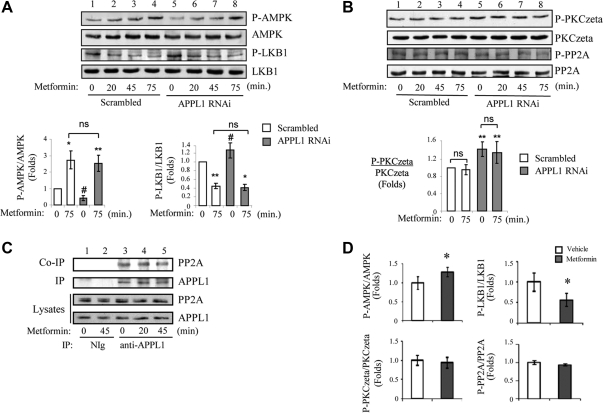
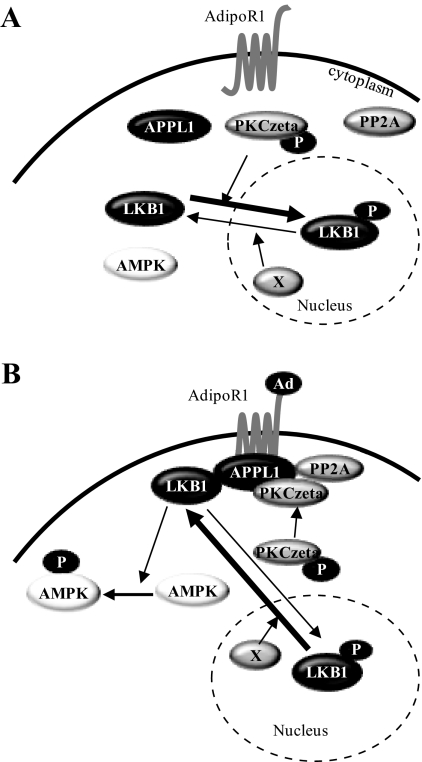
We recently found that the adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain and leucine zipper motif (APPL)1 is essential for mediating adiponectin signal to induce liver kinase B (LKB)1 cytosloic translocation, an essential step for activation of AMP-activated protein kinase (AMPK) in cells. However, the underlying molecular mechanisms remain unknown. Here, we demonstrate that treating C2C12 myotubes with adiponectin promoted APPL1 interaction with protein phosphatase 2A (PP2A) and protein kinase Cζ (PKCζ), leading to the activation of PP2A and subsequent dephosphorylation and inactivation of PKCζ. The adiponectin-induced inactivation of PKCζ results in dephosphorylation of LKB1 at Ser(307) and its subsequent translocation to the cytosol, where it stimulates AMPK activity. Interestingly, we found that metformin also induces LKB1 cytosolic translocation, but the stimulation is independent of APPL1 and the PP2A-PKCζ pathway. Together, our study uncovers a new mechanism underlying adiponectin-stimulated AMPK activation in muscle cells and shed light on potential targets for prevention and treatment of insulin resistance and its associated diseases.
Figures
References
-
- Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. 1995. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749 - PubMed
-
- Kadowaki T, Yamauchi T. 2005. Adiponectin and adiponectin receptors. Endocr Rev 26:439–451 - PubMed
-
- Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. 2010. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 464:1313–1319 - PubMed
-
- Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, Liu F, Dong LQ. 2006. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 8:516–523 - PubMed
