

A novel E2F-like protein involved in transcriptional activation of cyst wall protein genes in Giardia lamblia
- PMID: 21835923
- PMCID: PMC3190776
- DOI: 10.1074/jbc.M111.280206
A novel E2F-like protein involved in transcriptional activation of cyst wall protein genes in Giardia lamblia
Abstract
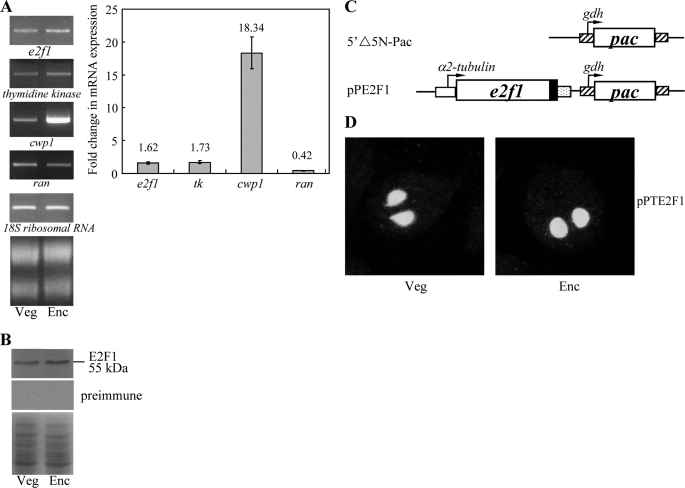
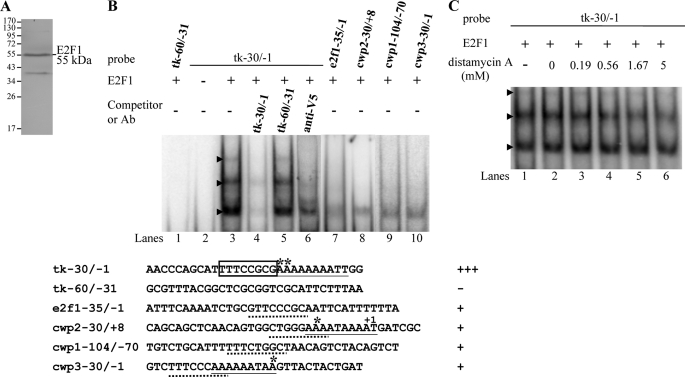
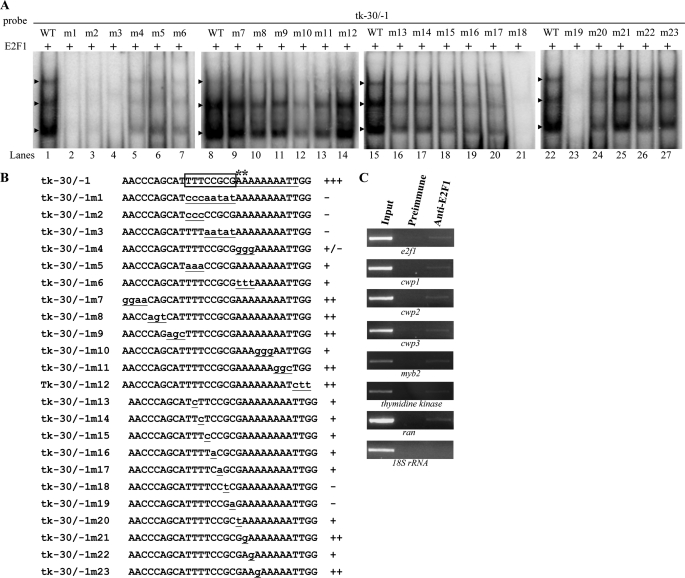
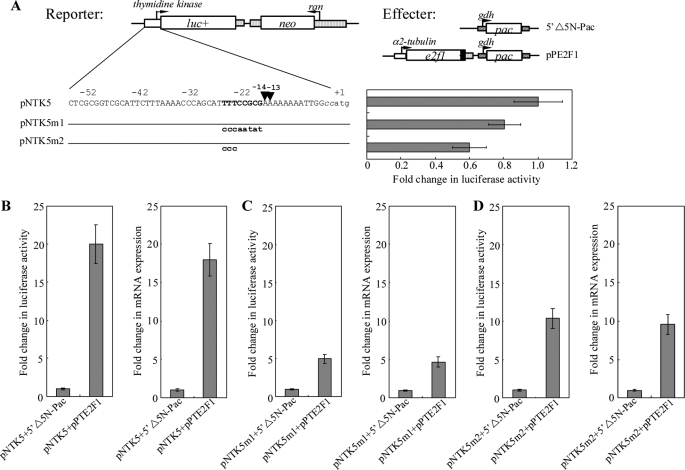
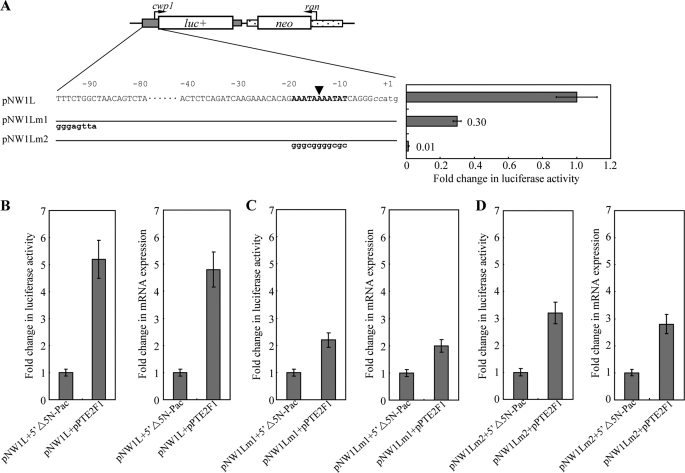
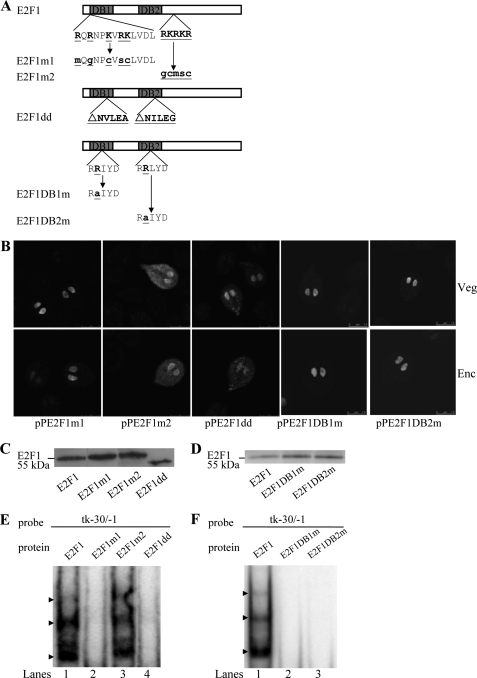
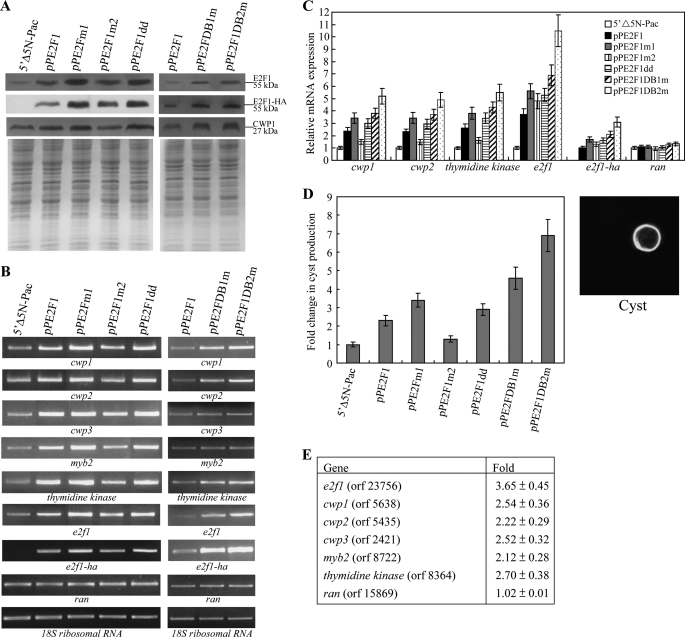
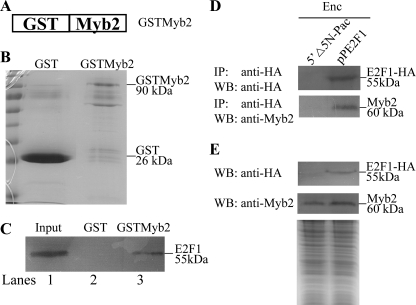
Giardia lamblia differentiates into resistant walled cysts for survival outside the host and transmission. During encystation, synthesis of cyst wall proteins is coordinately induced. The E2F family of transcription factors in higher eukaryotes is involved in cell cycle progression and cell differentiation. We asked whether Giardia has E2F-like genes and whether they influence gene expression during Giardia encystation. Blast searches of the Giardia genome database identified one gene (e2f1) encoding a putative E2F protein with two putative DNA-binding domains. We found that the e2f1 gene expression levels increased significantly during encystation. Epitope-tagged E2F1 was found to localize to nuclei. Recombinant E2F1 specifically bound to the thymidine kinase and cwp1-3 gene promoters. E2F1 contains several key residues for DNA binding, and mutation analysis revealed that its binding sequence is similar to those of the known E2F family proteins. The E2F1-binding sequences were positive cis-acting elements of the thymidine kinase and cwp1 promoters. We also found that E2F1 transactivated the thymidine kinase and cwp1 promoters through its binding sequences in vivo. Interestingly, E2F1 overexpression resulted in a significant increase of the levels of CWP1 protein, cwp1-3 gene mRNA, and cyst formation. We also found E2F1 can interact with Myb2, a transcription factor that coordinate up-regulates the cwp1-3 genes during encystation. Our results suggest that E2F family has been conserved during evolution and that E2F1 is an important transcription factor in regulation of the Giardia cwp genes, which are key to Giardia differentiation into cysts.
Figures

Similar articles
-
Functional redundancy of two Pax-like proteins in transcriptional activation of cyst wall protein genes in Giardia lamblia.PLoS One. 2012;7(2):e30614. doi: 10.1371/journal.pone.0030614. Epub 2012 Feb 15. PLoS One. 2012. PMID: 22355320 Free PMC article.
-
A Novel Multiprotein Bridging Factor 1-Like Protein Induces Cyst Wall Protein Gene Expression and Cyst Differentiation in Giardia lamblia.Int J Mol Sci. 2021 Jan 29;22(3):1370. doi: 10.3390/ijms22031370. Int J Mol Sci. 2021. PMID: 33573049 Free PMC article.
-
A novel ARID/Bright-like protein involved in transcriptional activation of cyst wall protein 1 gene in Giardia lamblia.J Biol Chem. 2007 Mar 23;282(12):8905-14. doi: 10.1074/jbc.M611170200. Epub 2007 Jan 23. J Biol Chem. 2007. PMID: 17244608
-
Encystation of Giardia lamblia: a model for other parasites.Curr Opin Microbiol. 2007 Dec;10(6):554-9. doi: 10.1016/j.mib.2007.09.011. Epub 2007 Nov 5. Curr Opin Microbiol. 2007. PMID: 17981075 Free PMC article. Review.
-
[Secreted proteins of Giardia duodenalis--characteristic and role in biology of the parasite].Wiad Parazytol. 2005;51(1):15-9. Wiad Parazytol. 2005. PMID: 16841684 Review. Polish.
Cited by
-
Regulation of a Myb transcription factor by cyclin-dependent kinase 2 in Giardia lamblia.J Biol Chem. 2012 Feb 3;287(6):3733-50. doi: 10.1074/jbc.M111.298893. Epub 2011 Dec 13. J Biol Chem. 2012. PMID: 22167200 Free PMC article.
-
DNA topoisomerase II is involved in regulation of cyst wall protein genes and differentiation in Giardia lamblia.PLoS Negl Trop Dis. 2013 May 16;7(5):e2218. doi: 10.1371/journal.pntd.0002218. Print 2013. PLoS Negl Trop Dis. 2013. PMID: 23696909 Free PMC article.
-
A myeloid leukemia factor homolog is involved in tolerance to stresses and stress-induced protein metabolism in Giardia lamblia.Biol Direct. 2023 Apr 24;18(1):20. doi: 10.1186/s13062-023-00378-6. Biol Direct. 2023. PMID: 37095576 Free PMC article.
-
A Novel Spo11 Homologue Functions as a Positive Regulator in Cyst Differentiation in Giardia lamblia.Int J Mol Sci. 2021 Nov 2;22(21):11902. doi: 10.3390/ijms222111902. Int J Mol Sci. 2021. PMID: 34769330 Free PMC article.
-
Functional redundancy of two Pax-like proteins in transcriptional activation of cyst wall protein genes in Giardia lamblia.PLoS One. 2012;7(2):e30614. doi: 10.1371/journal.pone.0030614. Epub 2012 Feb 15. PLoS One. 2012. PMID: 22355320 Free PMC article.
References
-
- Ankarklev J., Jerlström-Hultqvist J., Ringqvist E., Troell K., Svärd S. G. (2010) Nat. Rev. Microbiol. 8, 413–422 - PubMed
-
- Celiksöz A., Aciöz M., Değerli S., Cinar Z., Elaldi N., Erandaç M. (2005) Pediatr. Int. 47, 567–571 - PubMed
-
- Gillin F. D., Reiner D. S., McCaffery J. M. (1996) Annu. Rev. Microbiol. 50, 679–705 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
