

Aging and Cardiac Fibrosis
- PMID: 21837283
- PMCID: PMC3153299
Aging and Cardiac Fibrosis
Abstract
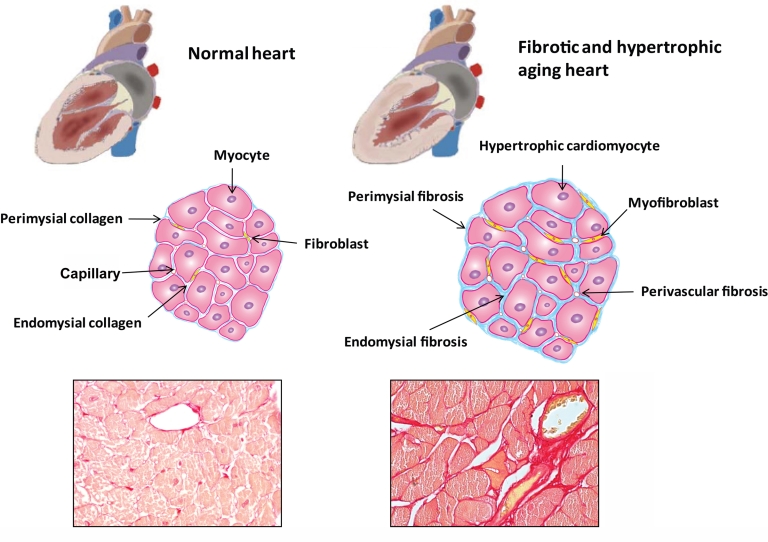
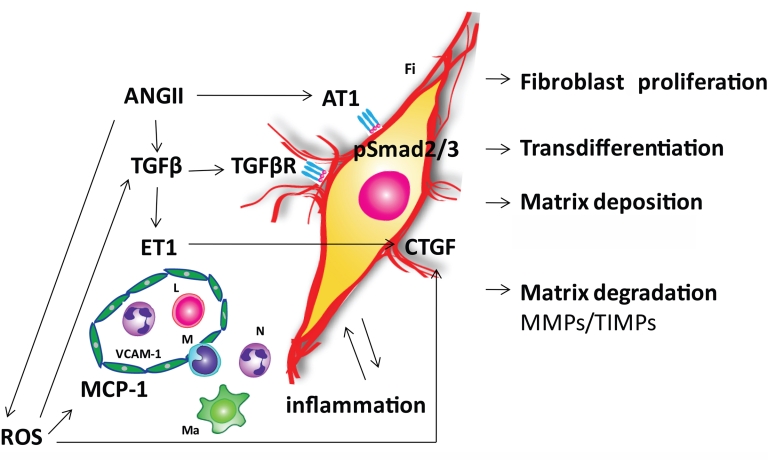
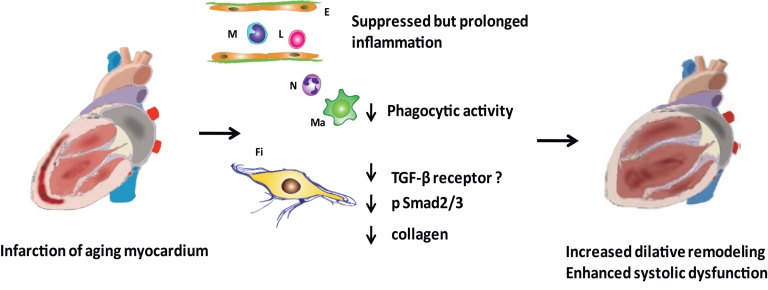
The aging heart is characterized by morphological and structural changes that lead to its functional decline and are associated with diminished ability to meet increased demand. Extensive evidence, derived from both clinical and experimental studies suggests that the aging heart undergoes fibrotic remodeling. Age-dependent accumulation of collagen in the heart leads to progressive increase in ventricular stiffness and impaired diastolic function. Increased mechanical load, due to reduced arterial compliance, and direct senescence-associated fibrogenic actions appear to be implicated in the pathogenesis of cardiac fibrosis in the elderly. Evolving evidence suggests that activation of several distinct molecular pathways may contribute to age-related fibrotic cardiac remodeling. Reactive oxygen species, chemokine-mediated recruitment of mononuclear cells and fibroblast progenitors, transforming growth factor (TGF)-β activation, endothelin-1 and angiotensin II signaling mediate interstitial and perivascular fibrosis in the senescent heart. Reduced collagen degradation may be more important than increased de novo synthesis in the pathogenesis of aging-associated fibrosis. In contrast to the baseline activation of fibrogenic pathways in the senescent heart, aging is associated with an impaired reparative response to cardiac injury and defective activation of reparative fibroblasts in response to growth factors. Because these reparative defects result in defective scar formation, senescent hearts are prone to adverse dilative remodeling following myocardial infarction. Understanding the pathogenesis of interstitial fibrosis in the aging heart and dissecting the mechanisms responsible for age-associated healing defects following cardiac injury are critical in order to design new strategies for prevention of adverse remodeling and heart failure in elderly patients.
Figures
References
-
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–54. - PubMed
-
- Kitzman DW, Gardin JM, Gottdiener JS, Arnold A, Boineau R, Aurigemma G, Marino EK, Lyles M, Cushman M, Enright PL. Importance of heart failure with preserved systolic function in patients > or = 65 years of age. CHS Research Group Cardiovascular Health Study. Am J Cardiol. 2001;87:413–9. - PubMed
-
- Vanoverschelde JJ, Essamri B, Vanbutsele R, d’Hondt A, Cosyns JR, Detry JR, Melin JA. Contribution of left ventricular diastolic function to exercise capacity in normal subjects. J Appl Physiol. 1993;74:2225–33. - PubMed
-
- Gagliano N, Arosio B, Santambrogio D, Balestrieri MR, Padoani G, Tagliabue J, Masson S, Vergani C, Annoni G. Age-dependent expression of fibrosis-related genes and collagen deposition in rat kidney cortex. J Gerontol A Biol Sci Med Sci. 2000;55:B365–72. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources