

Mechanisms of dendritic spine remodeling in a rat model of traumatic brain injury
- PMID: 21838518
- PMCID: PMC3261790
- DOI: 10.1089/neu.2011.1762
Mechanisms of dendritic spine remodeling in a rat model of traumatic brain injury
Abstract
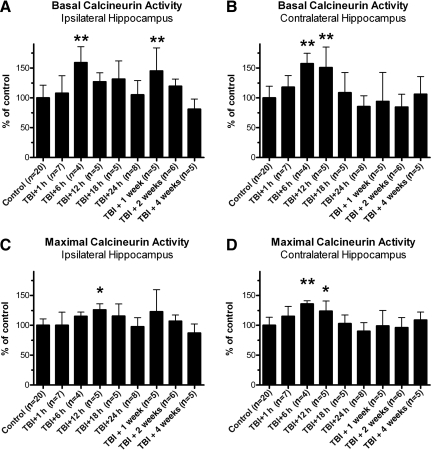
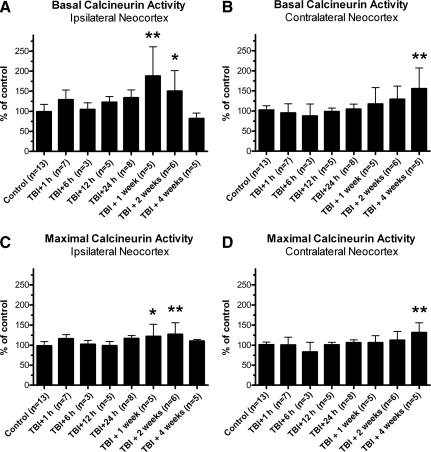
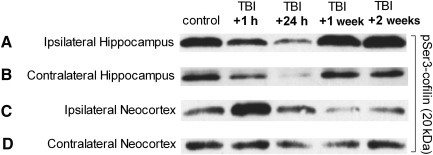
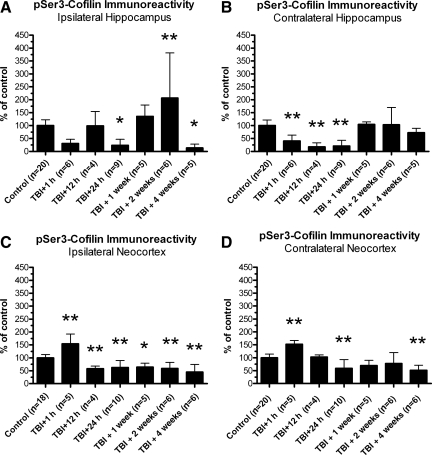
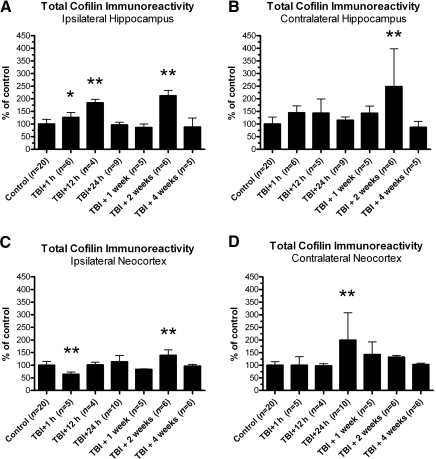
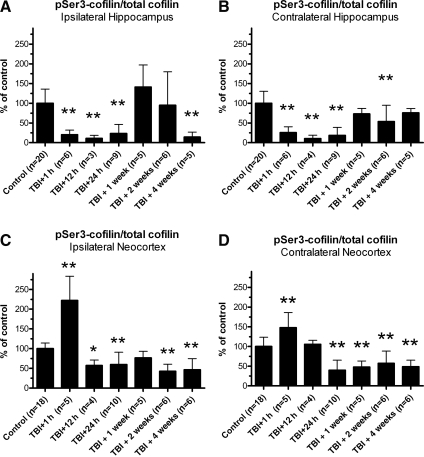
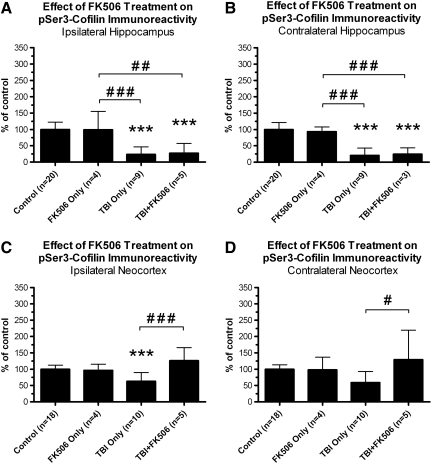
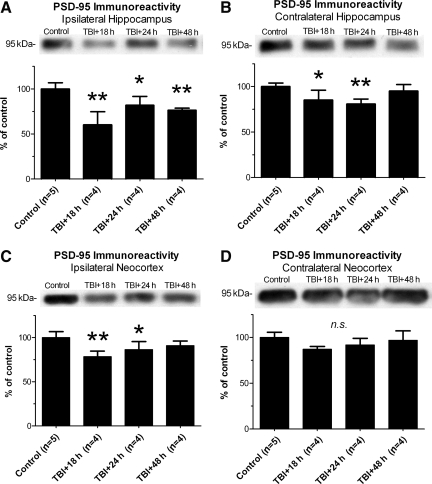
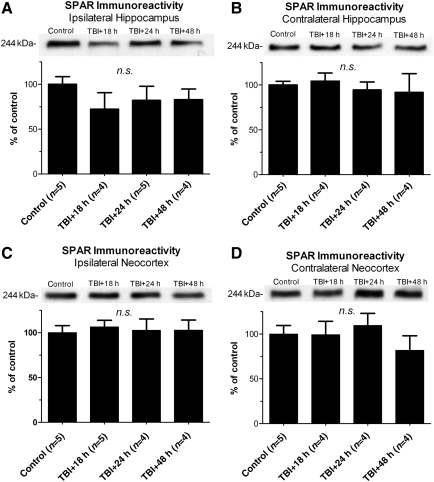
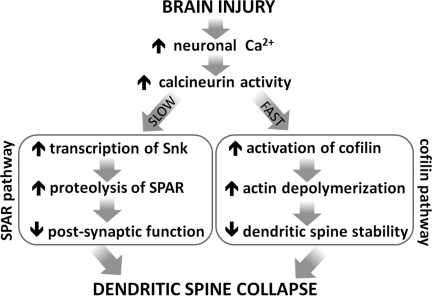
Traumatic brain injury (TBI), a leading cause of death and disability in the United States, causes potentially preventable damage in part through the dysregulation of neural calcium levels. Calcium dysregulation could affect the activity of the calcium-sensitive phosphatase calcineurin (CaN), with serious implications for neural function. The present study used both an in vitro enzymatic assay and Western blot analyses to characterize the effects of lateral fluid percussion injury on CaN activity and CaN-dependent signaling in the rat forebrain. TBI resulted in an acute alteration of CaN phosphatase activity and long-lasting alterations of its downstream effector, cofilin, an actin-depolymerizing protein. These changes occurred bilaterally in the neocortex and hippocampus, appeared to persist for hours after injury, and coincided with synapse degeneration, as suggested by a loss of the excitatory post-synaptic protein PSD-95. Interestingly, the effect of TBI on cofilin in some brain regions was blocked by a single bolus of the CaN inhibitor FK506, given 1 h post-TBI. Overall, these findings suggest a loss of synapse stability in both hemispheres of the laterally-injured brain, and offer evidence for region-specific, CaN-dependent mechanisms.
Figures
Similar articles
-
Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain.J Neurotrauma. 2012 Jan 20;29(2):201-17. doi: 10.1089/neu.2011.1761. J Neurotrauma. 2012. PMID: 21517673
-
Early, transient increase in complexin I and complexin II in the cerebral cortex following traumatic brain injury is attenuated by N-acetylcysteine.J Neurotrauma. 2006 Jan;23(1):86-96. doi: 10.1089/neu.2006.23.86. J Neurotrauma. 2006. PMID: 16430375
-
Blockade of Astrocytic Calcineurin/NFAT Signaling Helps to Normalize Hippocampal Synaptic Function and Plasticity in a Rat Model of Traumatic Brain Injury.J Neurosci. 2016 Feb 3;36(5):1502-15. doi: 10.1523/JNEUROSCI.1930-15.2016. J Neurosci. 2016. PMID: 26843634 Free PMC article.
-
Stress and trauma: BDNF control of dendritic-spine formation and regression.Prog Neurobiol. 2014 Jan;112:80-99. doi: 10.1016/j.pneurobio.2013.10.005. Epub 2013 Nov 6. Prog Neurobiol. 2014. PMID: 24211850 Review.
-
Amyloid beta: a putative intra-spinal microtubule-depolymerizer to induce synapse-loss or dentritic spine shortening in Alzheimer's disease.Ital J Anat Embryol. 2009 Apr-Sep;114(2-3):109-20. Ital J Anat Embryol. 2009. PMID: 20198823 Review.
Cited by
-
Pycnogenol protects CA3-CA1 synaptic function in a rat model of traumatic brain injury.Exp Neurol. 2016 Feb;276:5-12. doi: 10.1016/j.expneurol.2015.11.006. Epub 2015 Nov 29. Exp Neurol. 2016. PMID: 26607913 Free PMC article.
-
Preclinical Western Blot in the Era of Digital Transformation and Reproducible Research, an Eastern Perspective.Interdiscip Sci. 2021 Sep;13(3):490-499. doi: 10.1007/s12539-021-00442-7. Epub 2021 Jun 2. Interdiscip Sci. 2021. PMID: 34080131
-
Molecular mechanisms of cognitive dysfunction following traumatic brain injury.Front Aging Neurosci. 2013 Jul 9;5:29. doi: 10.3389/fnagi.2013.00029. eCollection 2013. Front Aging Neurosci. 2013. PMID: 23847533 Free PMC article.
-
The Nociceptin/Orphanin FQ peptide receptor antagonist, SB-612111, improves cerebral blood flow in a rat model of traumatic brain injury.Front Pharmacol. 2023 Oct 18;14:1272969. doi: 10.3389/fphar.2023.1272969. eCollection 2023. Front Pharmacol. 2023. PMID: 37920208 Free PMC article.
-
Xuefu zhuyu decoction improves cognitive impairment in experimental traumatic brain injury via synaptic regulation.Oncotarget. 2017 Jun 30;8(42):72069-72081. doi: 10.18632/oncotarget.18895. eCollection 2017 Sep 22. Oncotarget. 2017. PMID: 29069769 Free PMC article.
References
-
- Ansari M.A. Roberts K.N. Scheff S.W. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J. Neurotrauma. 2008b;25:513–526. - PubMed
-
- Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protien-dye binding. Anal. Biochem. 1976;72:248–254. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
