

JNK1, but not JNK2, is required in two mechanistically distinct models of inflammatory arthritis
- PMID: 21839715
- PMCID: PMC3181375
- DOI: 10.1016/j.ajpath.2011.06.019
JNK1, but not JNK2, is required in two mechanistically distinct models of inflammatory arthritis
Abstract
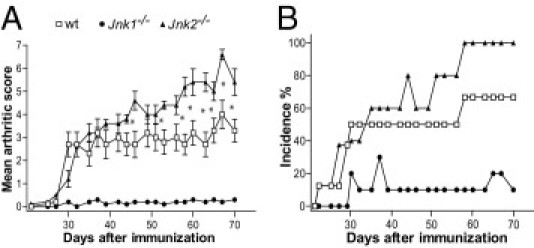
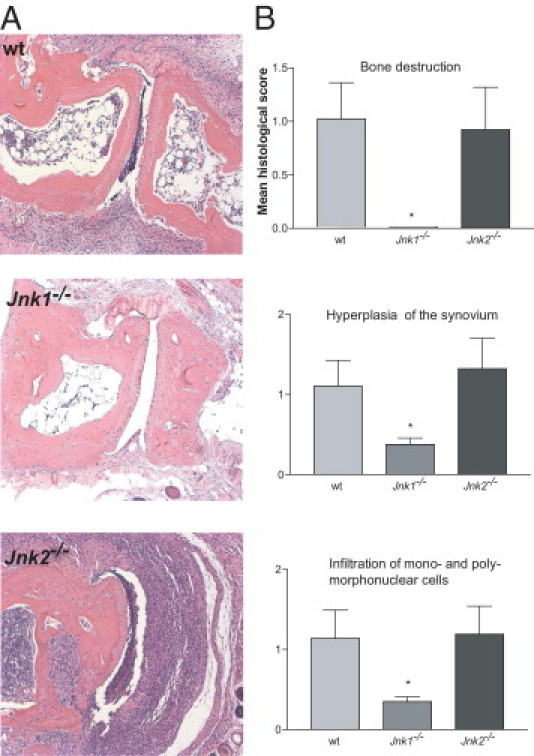
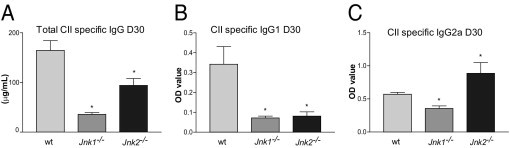
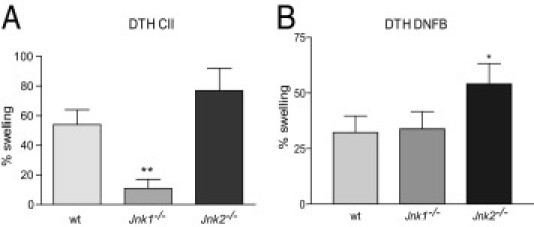
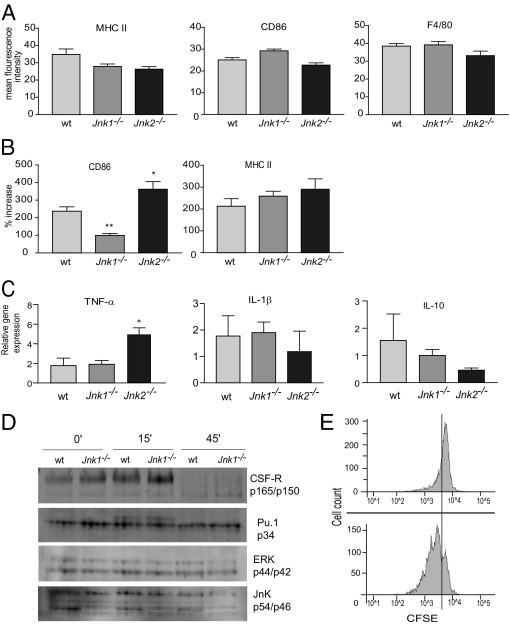
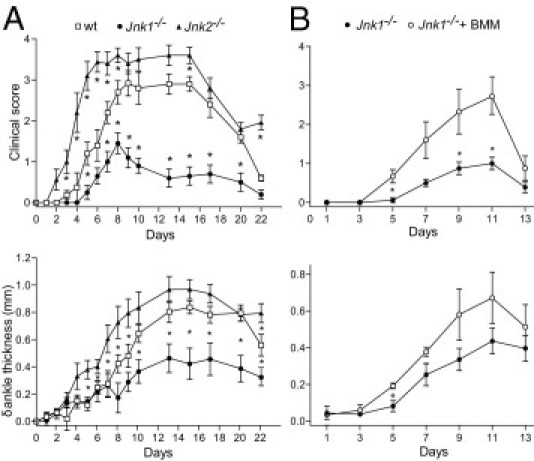
The roles of the c-Jun N-terminal kinases (JNKs) in inflammatory arthritis have been investigated; however, the roles of each isotype (ie, JNK1 and JNK2) in rheumatoid arthritis and conclusions about whether inhibition of one or both is necessary for amelioration of disease are unclear. By using JNK1- or JNK2-deficient mice in the collagen-induced arthritis and the KRN T-cell receptor transgenic mouse on C57BL/6 nonobese diabetic (K/BxN) serum transfer arthritis models, we demonstrate that JNK1 deficiency results in protection from arthritis, as judged by clinical score and histological evaluation in both models of inflammatory arthritis. In contrast, abrogation of JNK2 exacerbates disease. In collagen-induced arthritis, the distinct roles of the JNK isotypes can, at least in part, be explained by altered regulation of CD86 expression in JNK1- or JNK2-deficient macrophages in response to microbial products, thereby affecting T-cell-mediated immunity. The protection from K/BxN serum-induced arthritis in Jnk1(-/-) mice can also be explained by inept macrophage function because adoptive transfer of wild-type macrophages to Jnk1(-/-) mice restored disease susceptibility. Thus, our results provide a possible explanation for the modest therapeutic effects of broad JNK inhibitors and suggest that future therapies should selectively target the JNK1 isoform.
Copyright © 2011 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Schett G., Tohidast-Akrad M., Smolen J.S., Schmid B.J., Steiner C.W., Bitzan P., Zenz P., Redlich K., Xu Q., Steiner G. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000;43:2501–2512. - PubMed
-
- Han Z., Chang L., Yamanishi Y., Karin M., Firestein G.S. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. 2002;46:818–823. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
