

Genetic spectrum of hereditary neuropathies with onset in the first year of life
- PMID: 21840889
- PMCID: PMC3170533
- DOI: 10.1093/brain/awr184
Genetic spectrum of hereditary neuropathies with onset in the first year of life
Abstract
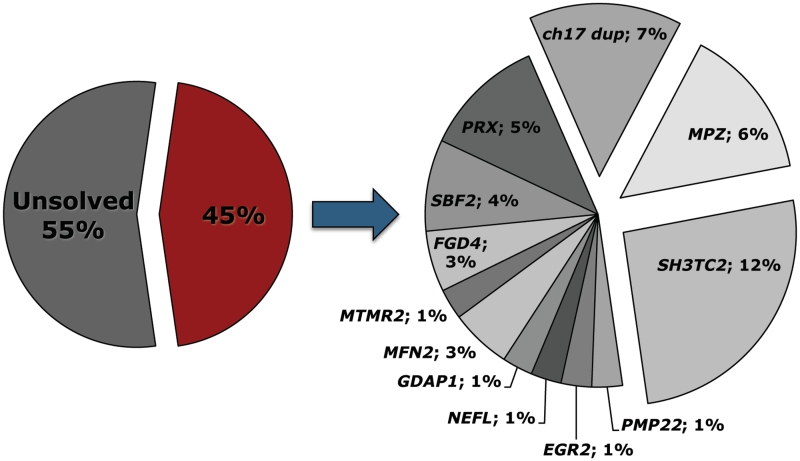
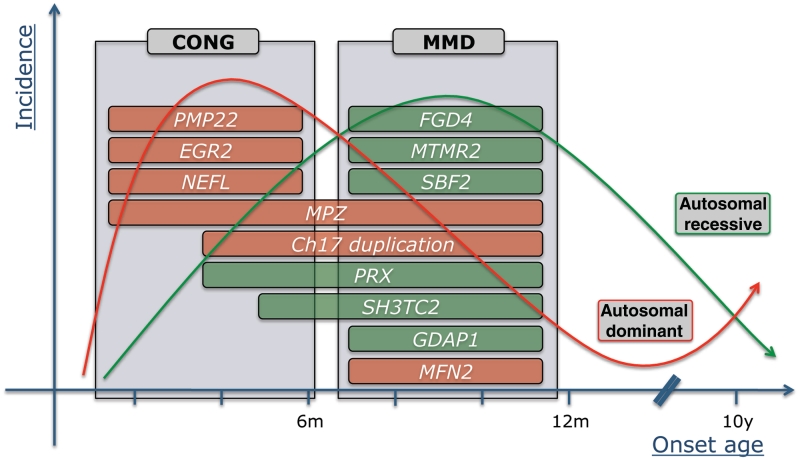
Early onset hereditary motor and sensory neuropathies are rare disorders encompassing congenital hypomyelinating neuropathy with disease onset in the direct post-natal period and Dejerine-Sottas neuropathy starting in infancy. The clinical spectrum, however, reaches beyond the boundaries of these two historically defined disease entities. De novo dominant mutations in PMP22, MPZ and EGR2 are known to be a typical cause of very early onset hereditary neuropathies. In addition, mutations in several other dominant and recessive genes for Charcot-Marie-Tooth disease may lead to similar phenotypes. To estimate mutation frequencies and to gain detailed insights into the genetic and phenotypic heterogeneity of early onset hereditary neuropathies, we selected a heterogeneous cohort of 77 unrelated patients who presented with symptoms of peripheral neuropathy within the first year of life. The majority of these patients were isolated in their family. We performed systematic mutation screening by means of direct sequencing of the coding regions of 11 genes: MFN2, PMP22, MPZ, EGR2, GDAP1, NEFL, FGD4, MTMR2, PRX, SBF2 and SH3TC2. In addition, screening for the Charcot-Marie-Tooth type 1A duplication on chromosome 17p11.2-12 was performed. In 35 patients (45%), mutations were identified. Mutations in MPZ, PMP22 and EGR2 were found most frequently in patients presenting with early hypotonia and breathing difficulties. The recessive genes FGD4, PRX, MTMR2, SBF2, SH3TC2 and GDAP1 were mutated in patients presenting with early foot deformities and variable delay in motor milestones after an uneventful neonatal period. Several patients displaying congenital foot deformities but an otherwise normal early development carried the Charcot-Marie-Tooth type 1A duplication. This study clearly illustrates the genetic heterogeneity underlying hereditary neuropathies with infantile onset.
Figures
References
-
- Azzedine H, Bolino A, Taieb T, Birouk N, Di Duca M, Bouhouche A, et al. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet. 2003;72:1141–53. - PMC - PubMed
-
- Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MA, Georgiou DM, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. 2000;25:17–9. - PubMed
-
- Burns J, Ryan MM, Ouvrier RA. Evolution of foot and ankle manifestations in children with CMT1A. Muscle Nerve. 2009;39:158–66. - PubMed
-
- Ceuterick-de Groote C, De Jonghe P, Timmerman V, Van Goethem G, Lofgren A, Ceulemans B, et al. Infantile demyelinating neuropathy associated with a de novo point mutation on Ser72 in PMP22 and basal lamina onion bulbs in skin biopsy. Pathol Res Pract. 2001;197:193–8. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
