

Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport
- PMID: 21841783
- PMCID: PMC3888087
- DOI: 10.1038/nm.2414
Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport
Abstract
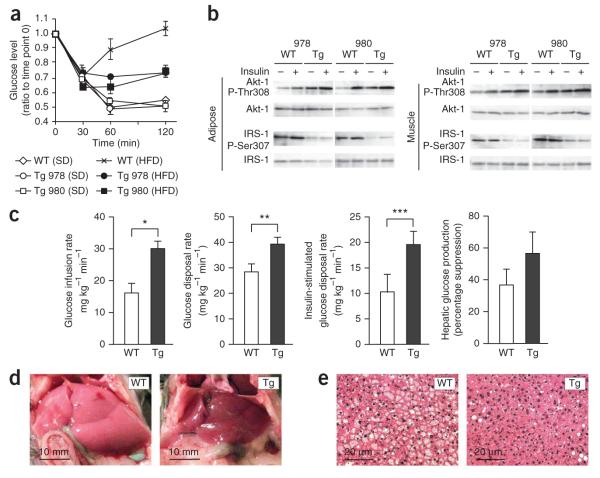
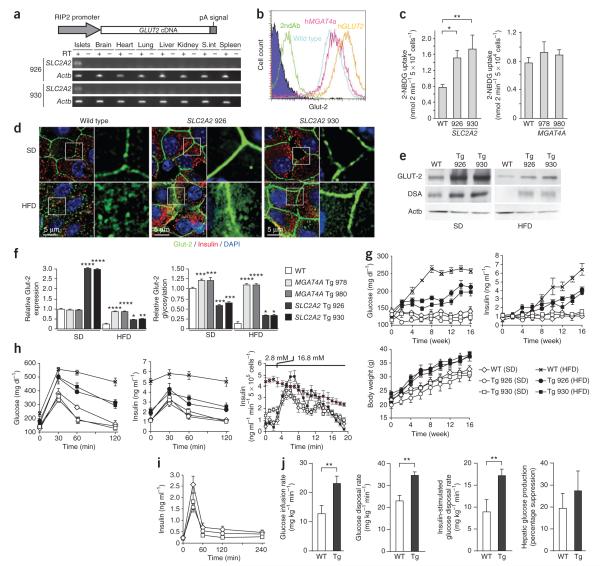
A connection between diet, obesity and diabetes exists in multiple species and is the basis of an escalating human health problem. The factors responsible provoke both insulin resistance and pancreatic beta cell dysfunction but remain to be fully identified. We report a combination of molecular events in human and mouse pancreatic beta cells, induced by elevated levels of free fatty acids or by administration of a high-fat diet with associated obesity, that comprise a pathogenic pathway to diabetes. Elevated concentrations of free fatty acids caused nuclear exclusion and reduced expression of the transcription factors FOXA2 and HNF1A in beta cells. This resulted in a deficit of GnT-4a glycosyltransferase expression in beta cells that produced signs of metabolic disease, including hyperglycemia, impaired glucose tolerance, hyperinsulinemia, hepatic steatosis and diminished insulin action in muscle and adipose tissues. Protection from disease was conferred by enforced beta cell-specific GnT-4a protein glycosylation and involved the maintenance of glucose transporter expression and the preservation of glucose transport. We observed that this pathogenic process was active in human islet cells obtained from donors with type 2 diabetes; thus, illuminating a pathway to disease implicated in the diet- and obesity-associated component of type 2 diabetes mellitus.
Figures
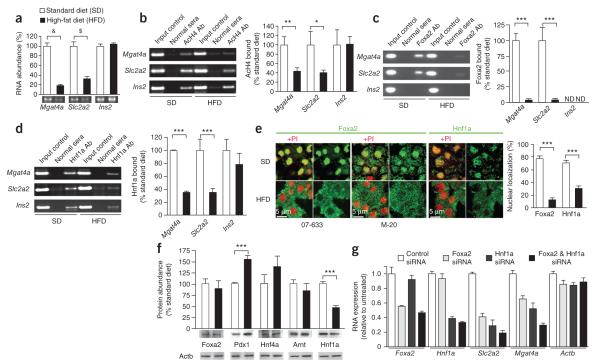
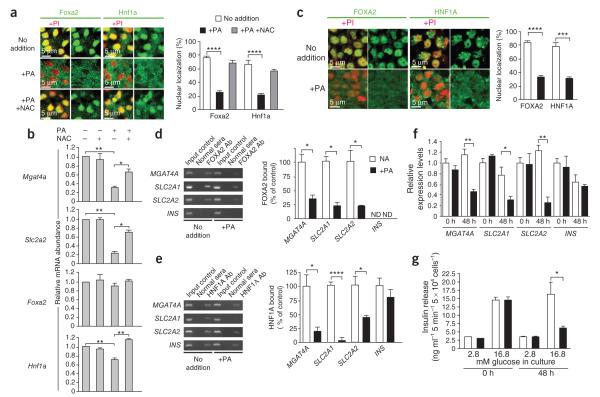
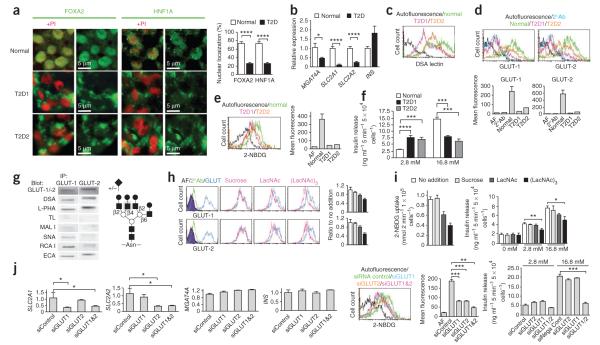
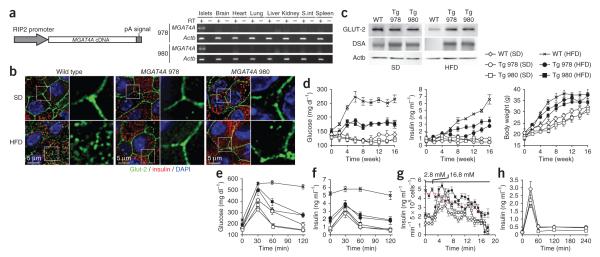
Comment in
-
Of fat, β cells, and diabetes.Cell Metab. 2011 Oct 5;14(4):439-40. doi: 10.1016/j.cmet.2011.09.006. Cell Metab. 2011. PMID: 21982703
References
-
- Olefsky JM, Courtney CH. Type 2 diabetes mellitus: etiology, pathogenesis and natural history. In: DeGroot LJ, et al., editors. Endocrinology. 5th edn W.B. Saunders; 2005. pp. 1093–1117.
-
- Weir GC, Leahy JL. Pathogenesis of non-insulin dependent (type II) diabetes mellitus. In: Joslin EP, Kahn CR, Weir GC, editors. Joslin’s Diabetes Mellitus. Lea and Febiger; 1994. pp. 240–264. Ch. 14.
-
- Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat. Med. 2006;12:75–80. - PubMed
-
- Korner J, Woods SC, Woodworth KA. Regulation of energy homeostasis and heath consequences in obesity. Am. J. Med. 2009;122:S12–S18. - PubMed
-
- Parillo M, Ricardi G. Diet composition and the risk of type 2 diabetes: epidemiological and clinical evidence. Br. J. Nutr. 2004;92:7–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DK033651/DK/NIDDK NIH HHS/United States
- P01 CA071932/CA/NCI NIH HHS/United States
- P01 DK074868/DK/NIDDK NIH HHS/United States
- R01 DK048247/DK/NIDDK NIH HHS/United States
- R01 GM100192/GM/NIGMS NIH HHS/United States
- R01 DK033651/DK/NIDDK NIH HHS/United States
- P30 DK063491/DK/NIDDK NIH HHS/United States
- T32 DK007494/DK/NIDDK NIH HHS/United States
- GM62116/GM/NIGMS NIH HHS/United States
- P50 HD012303/HD/NICHD NIH HHS/United States
- DK48247/DK/NIDDK NIH HHS/United States
- U54 HD 012303-25/HD/NICHD NIH HHS/United States
- CA71932/CA/NCI NIH HHS/United States
- DK063491/DK/NIDDK NIH HHS/United States
- DK074868/DK/NIDDK NIH HHS/United States
- U54 GM062116/GM/NIGMS NIH HHS/United States
- U54 HD012303/HD/NICHD NIH HHS/United States
- R37 DK033651/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
