

Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers
- PMID: 21842473
- PMCID: PMC3397249
- DOI: 10.1002/adma.201102313
Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers
Abstract
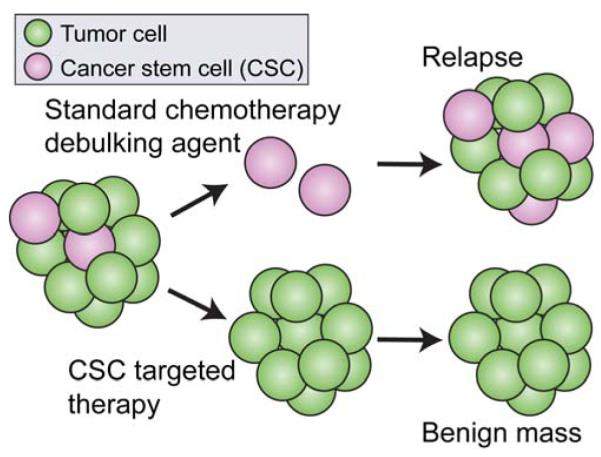
Cancer nanotheranostics aims to combine imaging and therapy of cancer through use of nanotechnology. The ability to engineer nanomaterials to interact with cancer cells at the molecular level can significantly improve the effectiveness and specificity of therapy to cancers that are currently difficult to treat. In particular, metastatic cancers, drug-resistant cancers, and cancer stem cells impose the greatest therapeutic challenge for targeted therapy. Targeted therapy can be achieved with appropriately designed drug delivery vehicles such as nanoparticles, adult stem cells, or T cells in immunotherapy. In this article, we first review the different types of nanotheranostic particles and their use in imaging, followed by the biological barriers they must bypass to reach the target cancer cells, including the blood, liver, kidneys, spleen, and particularly the blood-brain barrier. We then review how nanotheranostics can be used to improve targeted delivery and treatment of cancer cells. Finally, we discuss development of nanoparticles to overcome current limitations in cancer therapy.
Copyright © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures
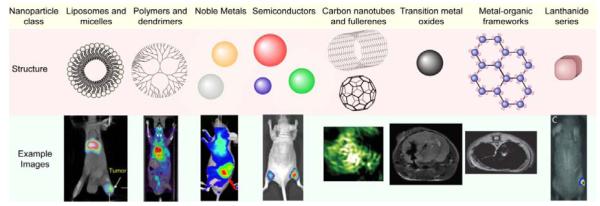
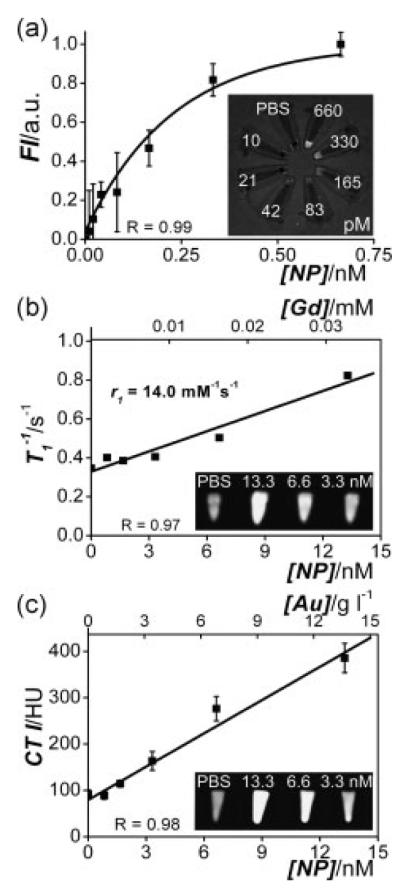
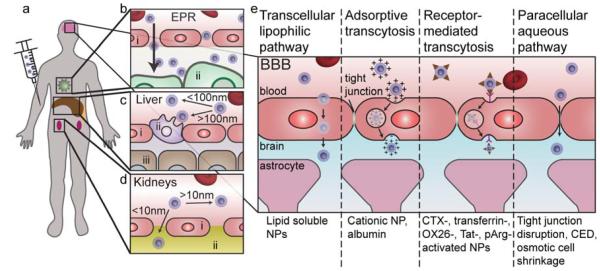
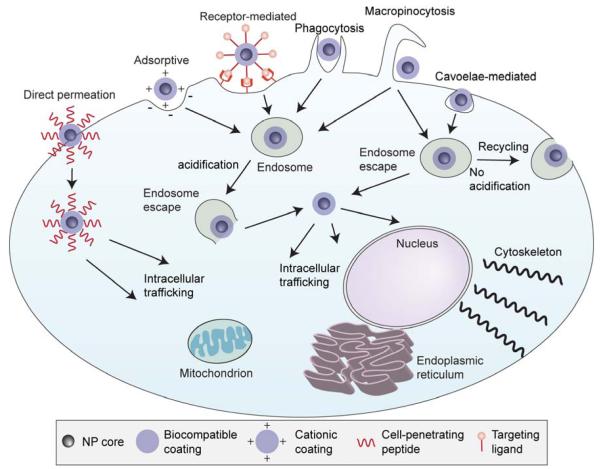
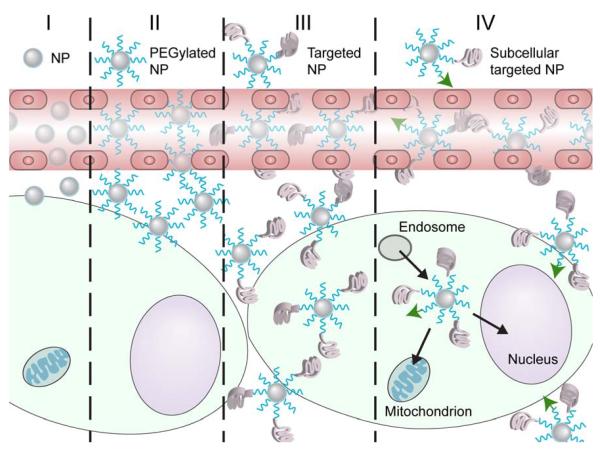
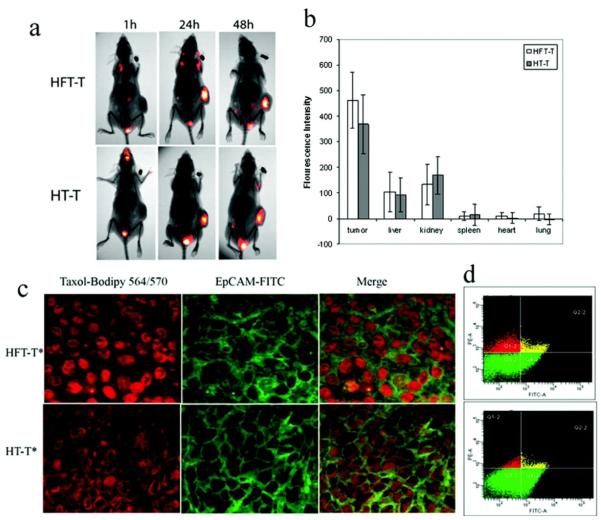
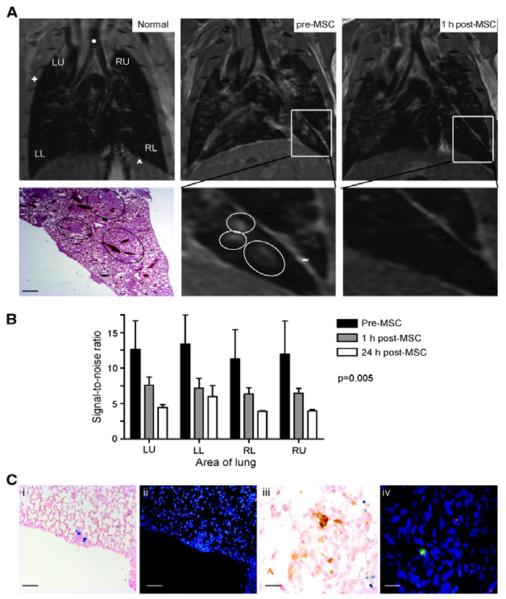
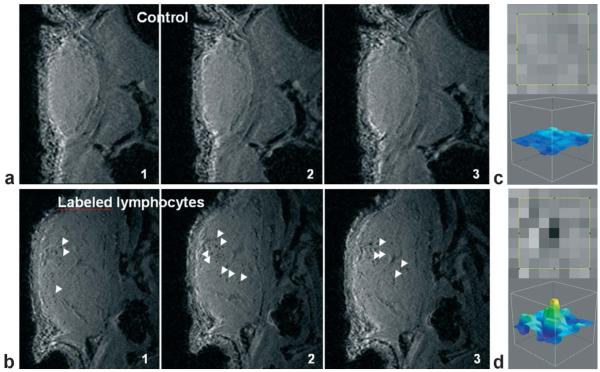
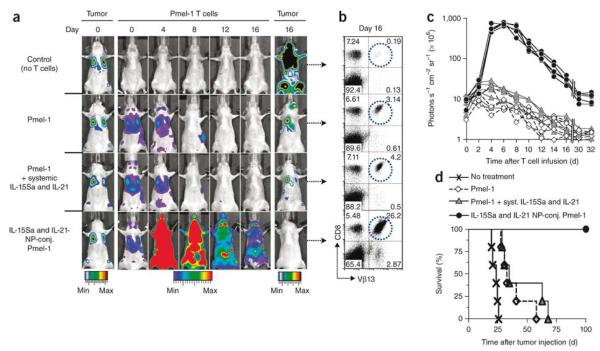
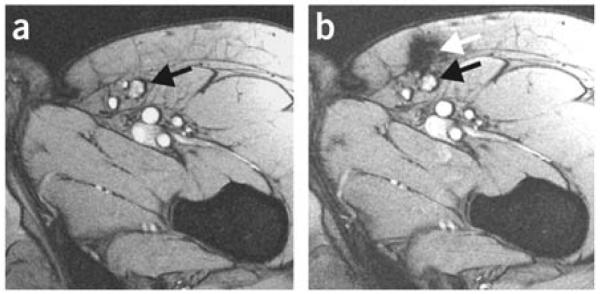
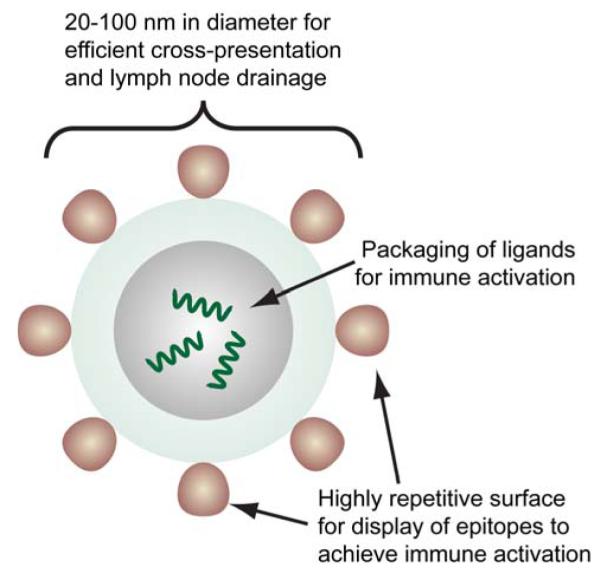
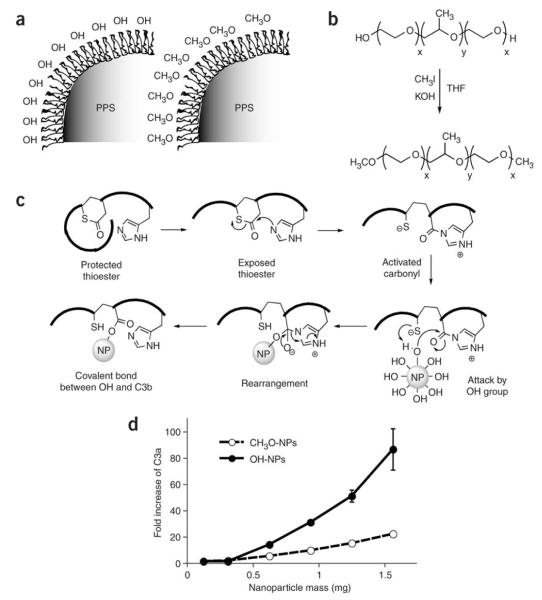
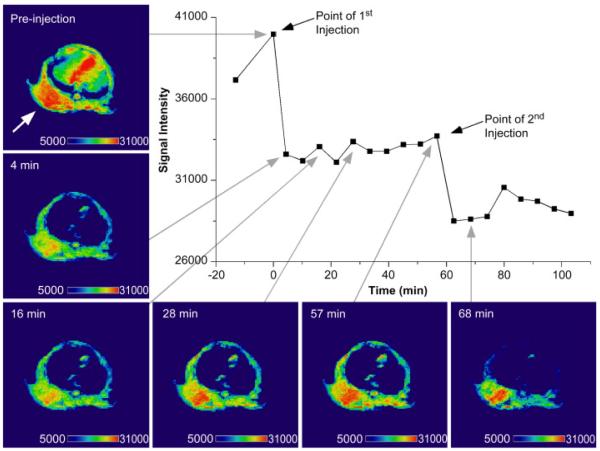
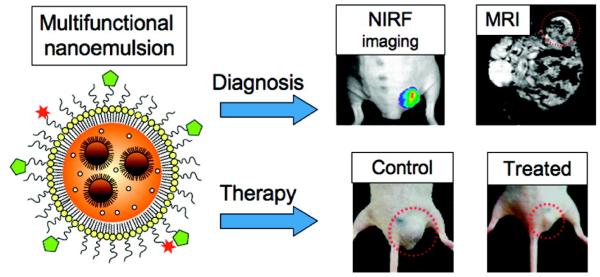
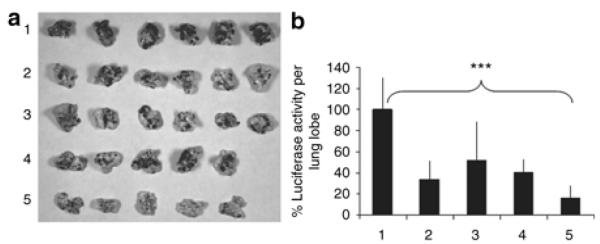
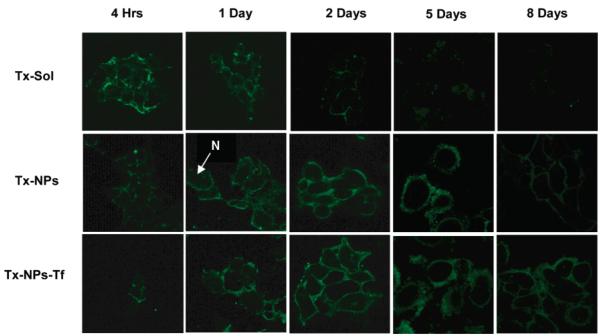
References
-
- World Health Organization; Geneva: 2011.
-
- Mehlen P, Puisieux A. Nat Rev Cancer. 2006;6:449. - PubMed
-
- Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Nat Rev Drug Discov. 2006;5:219. - PubMed
-
- Cho KJ, Wang X, Nie SM, Chen Z, Shin DM. Clin. Cancer Res. 2008;14:1310. - PubMed
-
- Brindle K. Nat. Rev. Cancer. 2008;8:94. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
