

Proteostasis and movement disorders: Parkinson's disease and amyotrophic lateral sclerosis
- PMID: 21844169
- PMCID: PMC3179340
- DOI: 10.1101/cshperspect.a007500
Proteostasis and movement disorders: Parkinson's disease and amyotrophic lateral sclerosis
Abstract
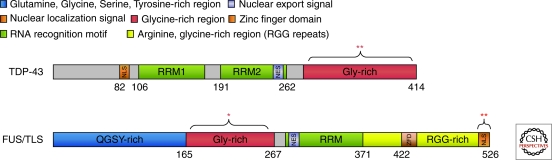
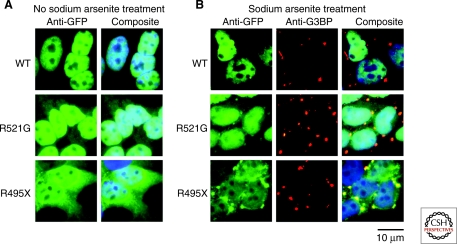
Parkinson's disease (PD) is a movement disorder that afflicts over one million in the U.S.; amyotrophic lateral sclerosis (ALS or Lou Gehrig's disease) is less prevalent but also has a high incidence. The two disorders sometimes present together, making a comparative study of interest. Both ALS and PD are neurodegenerative diseases, and are characterized by the presence of intraneuronal inclusions; however, different classes of neurons are affected and the primary protein in the inclusions differs between the diseases, and in some cases is different in distinct forms of the same disease. These observations might suggest that the more general approach of proteostasis pathway alteration would be a powerful one in treating these disorders. Examining results from human genetics and studies in model organisms, as well as from biochemical and biophysical characterization of the proteins involved in both diseases, we find that most instances of PD can be considered as arising from the misfolding, and self-association to a toxic species, of the small neuronal protein α-synuclein, and that proteostasis strategies are likely to be of value for this disorder. For ALS, the situation is much more complex and less clear-cut; the available data are most consistent with a view that ALS may actually be a family of disorders, presenting similarly but arising from distinct and nonoverlapping causes, including mislocalization of some properly folded proteins and derangement of RNA quality control pathways. Applying proteostasis approaches to this disease may require rethinking or broadening the concept of what proteostasis means.
Figures
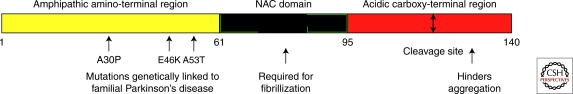
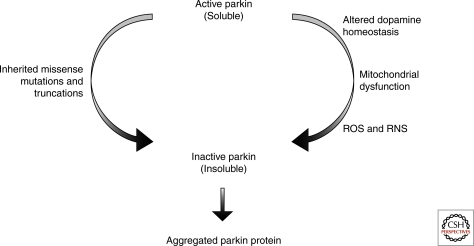
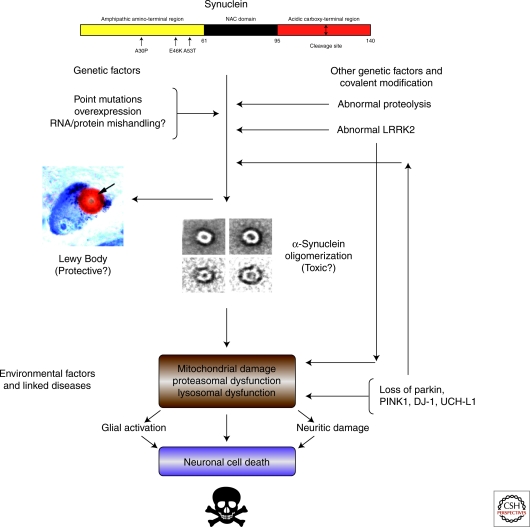
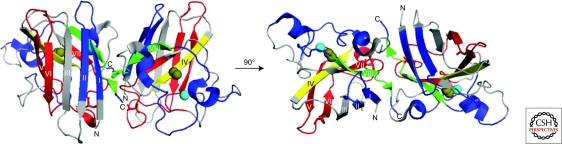
References
-
- Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. 2000. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: 239–252 - PubMed
-
- Anderson P, Kedersha N 2008. Stress granules: The Tao of RNA triage. Trends Biochem Sci 33: 141–150 - PubMed
-
- Andersson FI, Werrell EF, McMorran L, Crone WJ, Das C, Hsu ST, Jackson SE 2011. The effect of Parkinson’s-Disease-associated mutations on the deubiquitinating enzyme UCH-L1. J Mol Biol 407: 261–272 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous