

OatA, a peptidoglycan O-acetyltransferase involved in Listeria monocytogenes immune escape, is critical for virulence
- PMID: 21844299
- PMCID: PMC3156107
- DOI: 10.1093/infdis/jir396
OatA, a peptidoglycan O-acetyltransferase involved in Listeria monocytogenes immune escape, is critical for virulence
Abstract
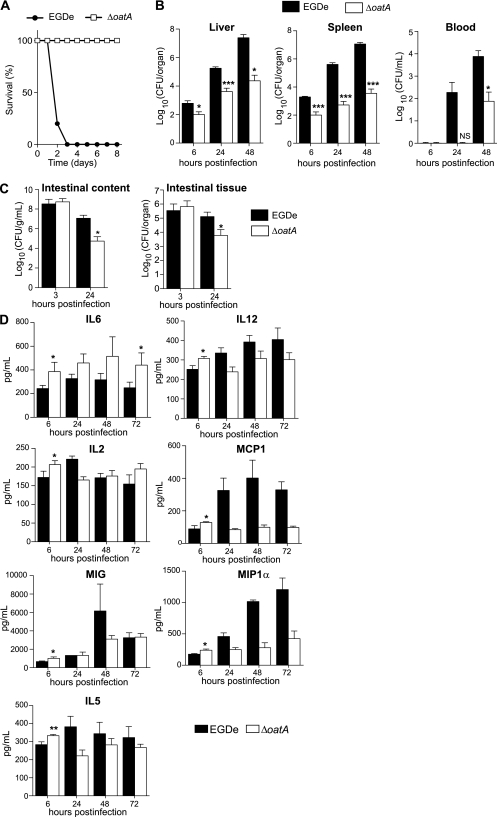
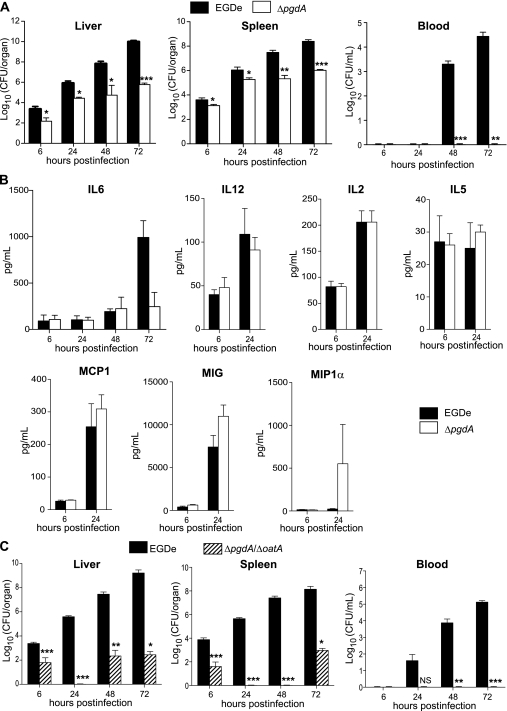
Microbial pathogens have evolved mechanisms to overcome immune responses and successfully infect their host. Here, we studied how Listeria monocytogenes evades immune detection by peptidoglycan (PGN) modification. By analyzing L. monocytogenes muropeptides, we detected O-acetylated muramic acid residues. We identified an O-acetyltransferase gene, oatA, in the L. monocytogenes genome sequence. Comparison of PGN from parental and isogenic oatA mutant strains showed that the O-acetyltransferase OatA O-acetylates Listeria PGN. We also found that PGN O-acetylation confers resistance to different types of antimicrobial compounds targeting bacterial cell wall such as lysozyme, β-lactam antibiotics, and bacteriocins and that O-acetylation is required for Listeria growth in macrophages. Moreover, oatA mutant virulence is drastically affected in mice following intravenous or oral inoculation. In addition, the oatA mutant induced early secretion of proinflammatory cytokines and chemokines in vivo. These results suggest an important role for OatA in limiting innate immune responses and promoting bacterial survival in the infected host.
Figures


References
-
- Pizarro-Cerda J, Cossart P. Listeria monocytogenes membrane trafficking and lifestyle: the exception or the rule? Annu Rev Cell Dev Biol. 2009;25:649–70. - PubMed
-
- Hamon M, Bierne H, Cossart P. Listeria monocytogenes: a multifaceted model. Nat Rev Microbiol. 2006;4:423–34. - PubMed
-
- Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–23. - PubMed
-
- Stavru F, Archambaud C, Cossart P. Cell biology and immunology of Listeria monocytogenes infections: novel insights. Immunol Rev. 2011;240:160–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
