

The origins and evolution of freeze-etch electron microscopy
- PMID: 21844598
- PMCID: PMC3202940
- DOI: 10.1093/jmicro/dfr044
The origins and evolution of freeze-etch electron microscopy
Abstract
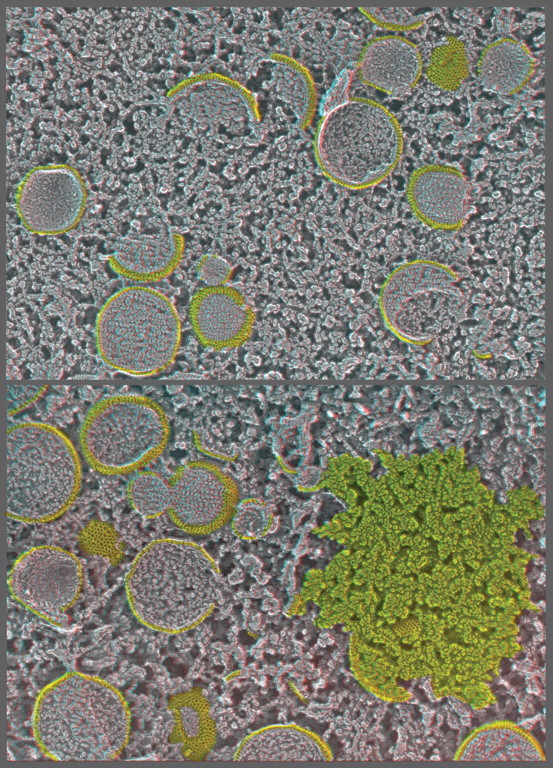
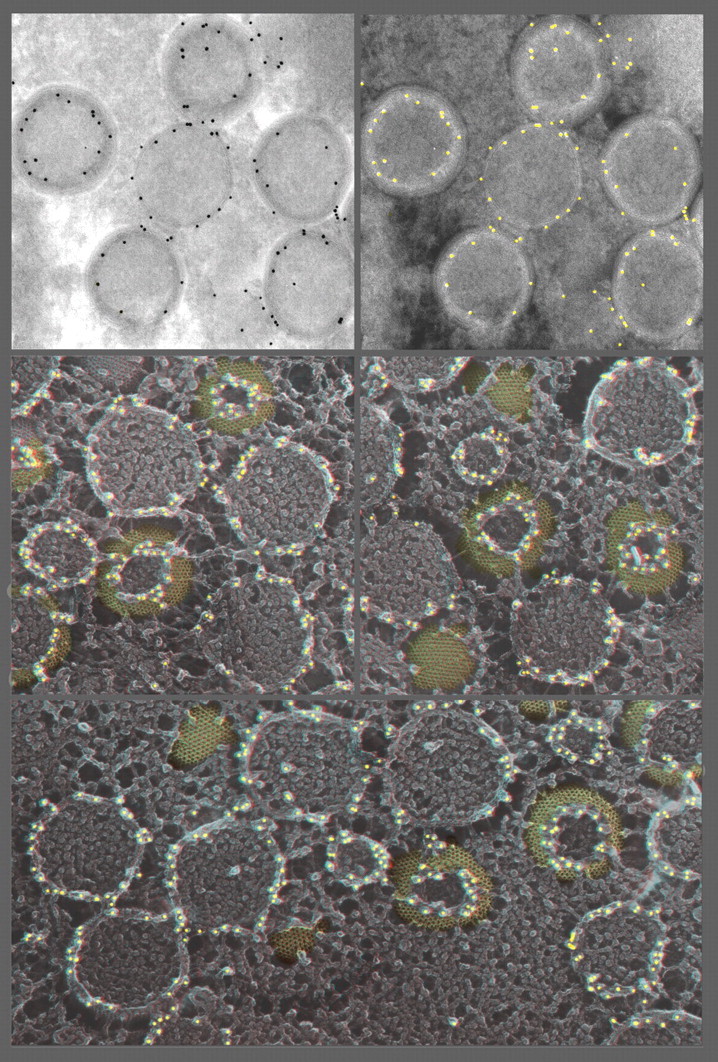
The introduction of the Balzers freeze-fracture machine by Moor in 1961 had a much greater impact on the advancement of electron microscopy than he could have imagined. Devised originally to circumvent the dangers of classical thin-section techniques, as well as to provide unique en face views of cell membranes, freeze-fracturing proved to be crucial for developing modern concepts of how biological membranes are organized and proved that membranes are bilayers of lipids within which proteins float and self-assemble. Later, when freeze-fracturing was combined with methods for freezing cells that avoided the fixation and cryoprotection steps that Moor still had to use to prepare the samples for his original invention, it became a means for capturing membrane dynamics on the millisecond time-scale, thus allowing a deeper understanding of the functions of biological membranes in living cells as well as their static ultrastructure. Finally, the realization that unfixed, non-cryoprotected samples could be deeply vacuum-etched or even freeze-dried after freeze-fracturing opened up a whole new way to image all the other molecular components of cells besides their membranes and also provided a powerful means to image the interactions of all the cytoplasmic components with the various membranes of the cell. The purpose of this review is to outline the history of these technical developments, to describe how they are being used in electron microscopy today and to suggest how they can be improved in order to further their utility for biological electron microscopy in the future.
Figures

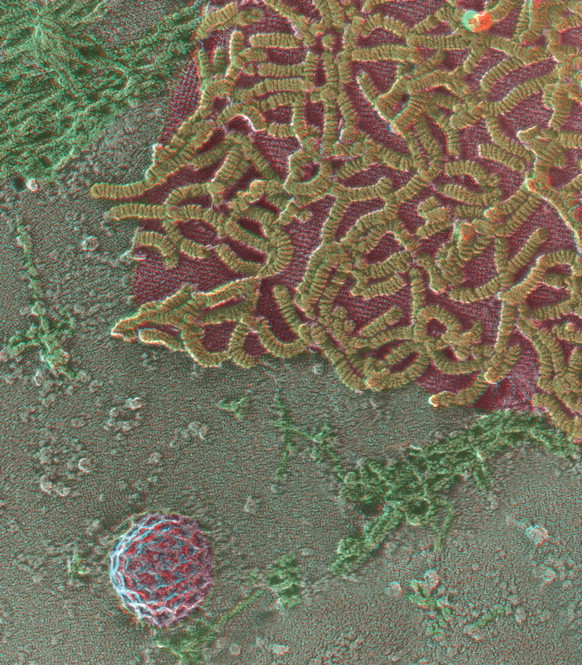
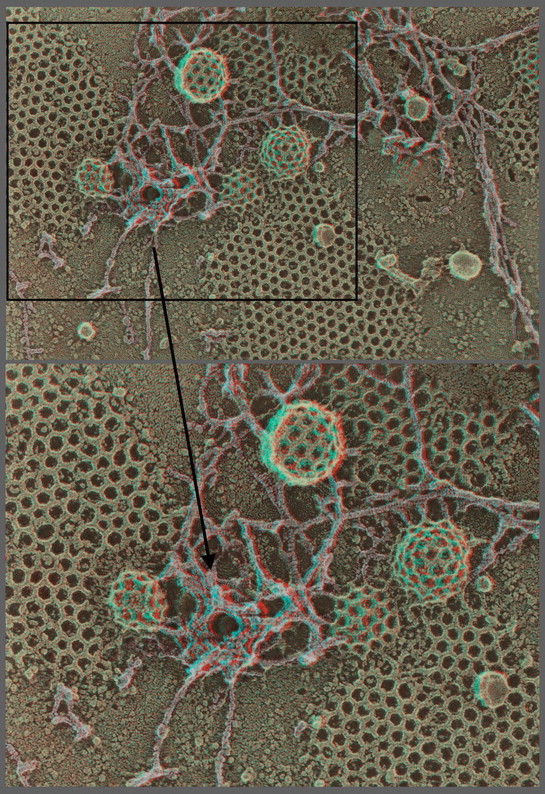
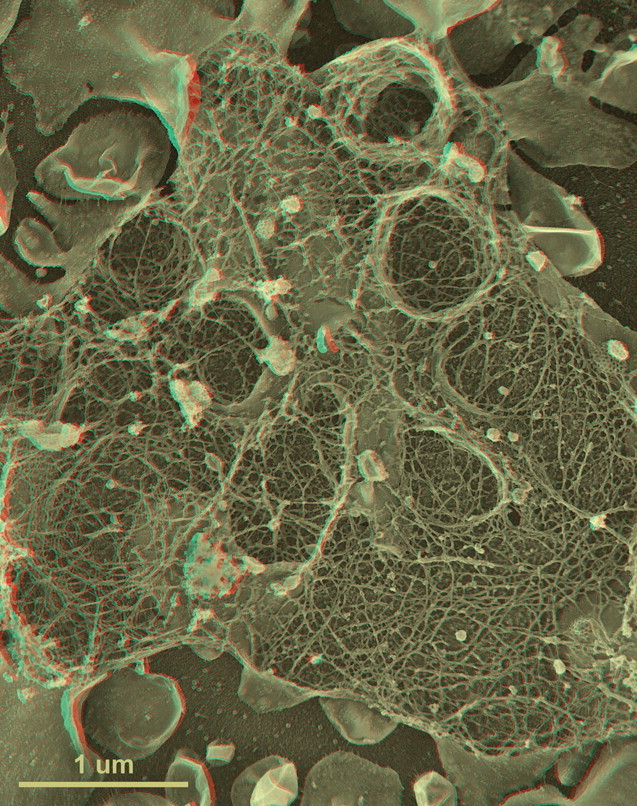

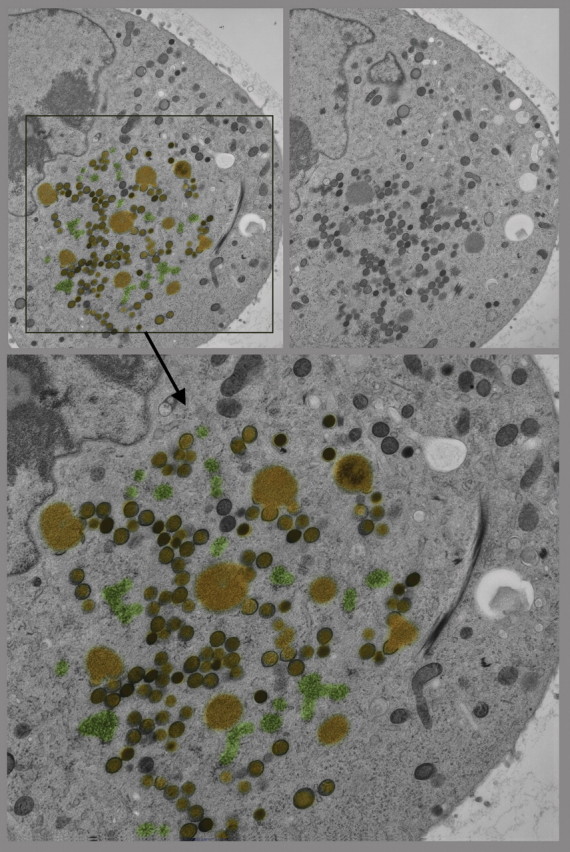
References
-
- Moor H, Muhlethaler K, Waldner H, Frey-Wyssling A. A new freezing-ultramicrotome. J. Biophys. Biochem. Cytol. 1961;10:1–13. doi:10.1083/jcb.10.1.1. - DOI - PMC - PubMed
-
- Heuser J E, Reese T S, Landis D M. Preservation of synaptic structure by rapid freezing. Cold Spring Harb. Symp. Quant. Biol. 1976;40:17–24. - PubMed
-
- Heuser J E, Reese T S, Dennis M J, Jan Y, Jan L, Evans L. Synaptic vesicle exocytosis captured by quick freezing and correlated with quantal transmitter release. J. Cell. Biol. 1979;81:275–300. doi:10.1083/jcb.81.2.275. - DOI - PMC - PubMed
-
- Heuser J E, Salpeter S R. Organization of acetylcholine receptors in quick-frozen, deep-etched, and rotary-replicated Torpedo postsynaptic membrane. J. Cell. Biol. 1979;82:150–173. doi:10.1083/jcb.82.1.150. - DOI - PMC - PubMed
-
- Heuser J. Three-dimensional visualization of coated vesicle formation in fibroblasts. J. Cell. Biol. 1980;84:560–583. doi:10.1083/jcb.84.3.560. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
