

Near-atomic-resolution cryo-EM for molecular virology
- PMID: 21845206
- PMCID: PMC3155204
- DOI: 10.1016/j.coviro.2011.05.019
Near-atomic-resolution cryo-EM for molecular virology
Abstract
Electron cryo-microscopy (cryo-EM) is a technique in structural biology that is widely used to solve the three-dimensional structures of macromolecular assemblies, close to their biological and solution conditions. Recent improvements in cryo-EM and single-particle reconstruction methodologies have led to the determination of several virus structures at near-atomic resolution (3.3 - 4.6 Å). These cryo-EM structures not only resolve the Cα backbones and side-chain densities of viral capsid proteins, but also suggest functional roles that the protein domains and some key amino acid residues play. This paper reviews the recent advances in near-atomic-resolution cryo-EM for probing the mechanisms of virus assembly and morphogenesis.
Figures
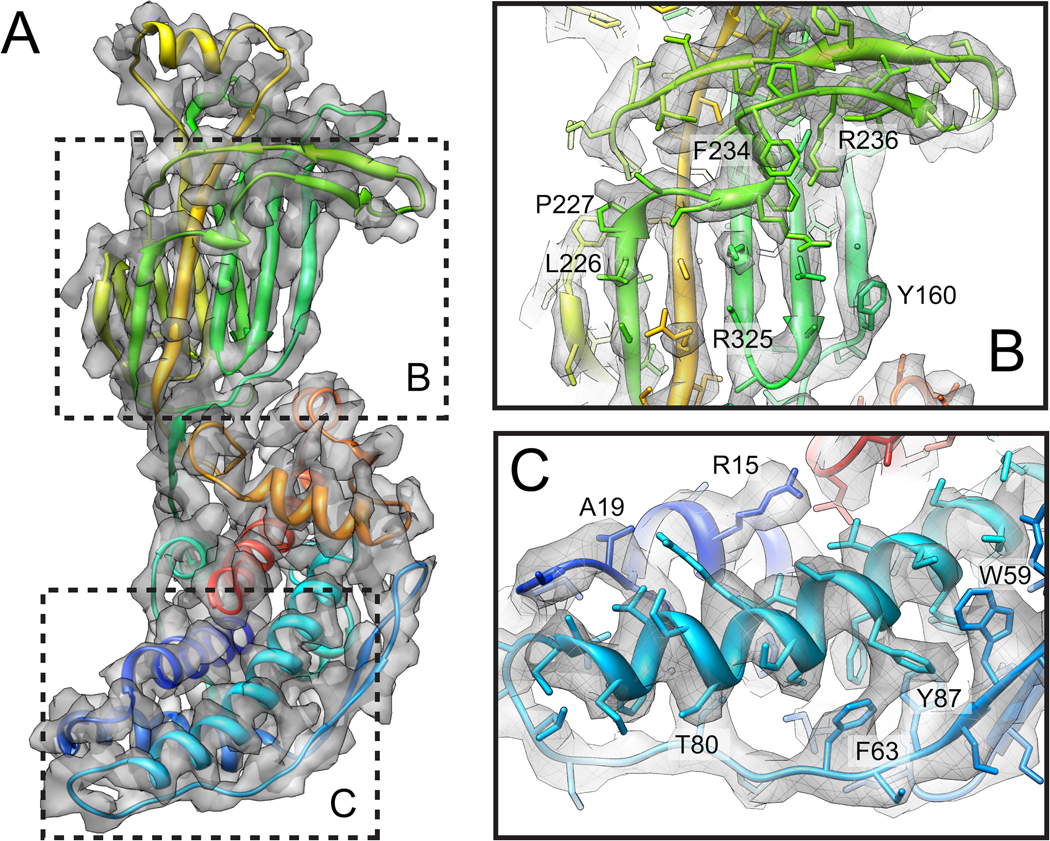
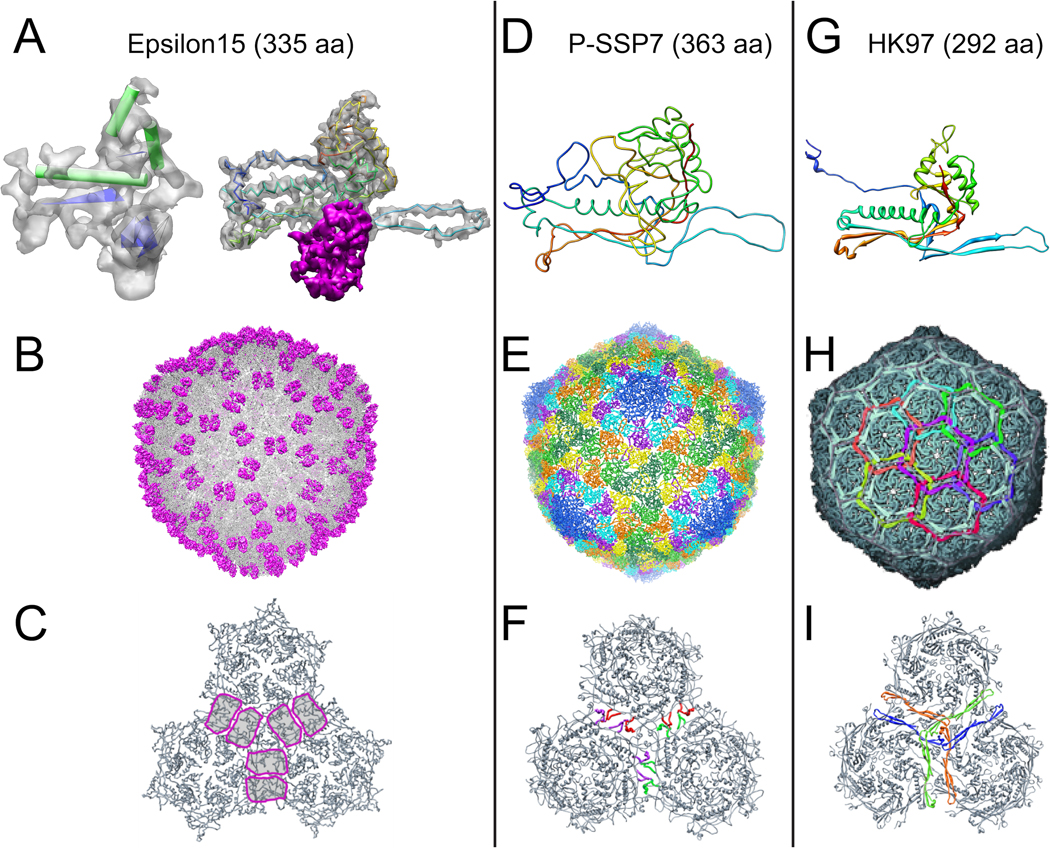
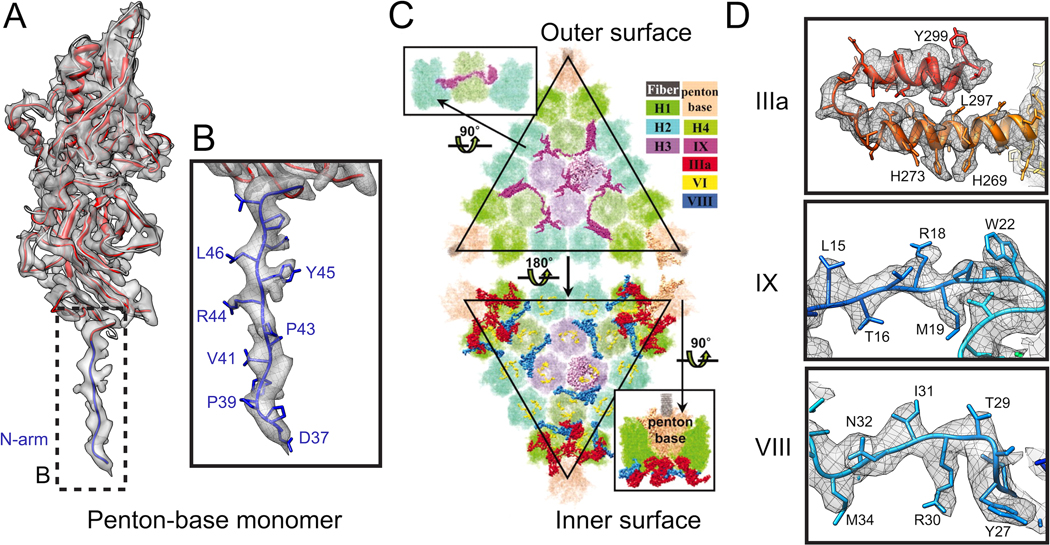
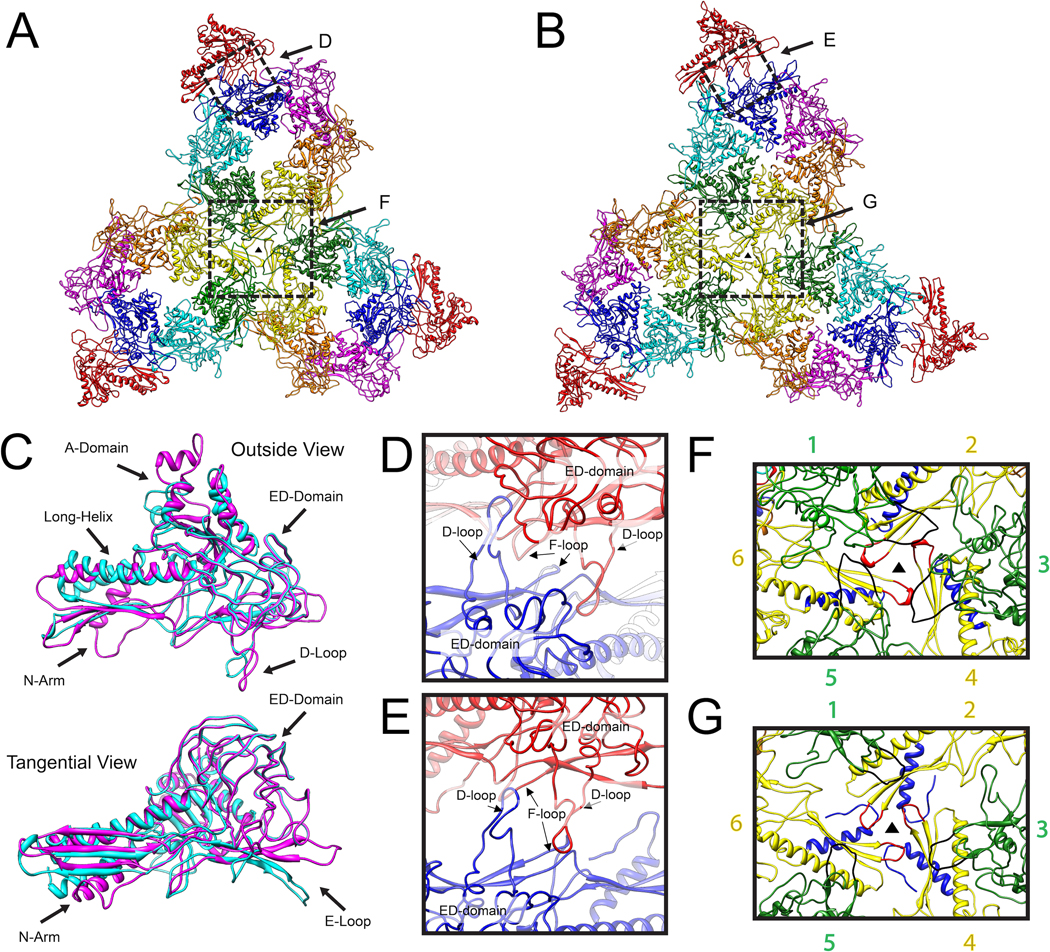
References
-
- Harrison SC. Principles of virus structure. Fields virology. 2001;1:53–85.
-
- Adrian M, Dubochet J, Lepault J, McDowall AW. Cryo-electron microscopy of viruses. Nature. 1984;308:32–36. - PubMed
-
- Glaeser RM. Electron crystallography of biological macromolecules. USA: Oxford University Press; 2007.
-
- De Rosier DJ, Klug A. Reconstruction of three dimensional structures from electron micrographs. Nature. 1968;217:130–134. - PubMed
-
-
Chen DH, Baker ML, Hryc CF, Dimaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, et al. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc Natl Acad Sci U S A. 2011;108:1355–1360. Cryo-EM was used to resolve two morphogenetic states of the P22 phage at near-atomic resolution, revealing detailed conformational changes upon maturation. A virus assembly mechanism was proposed based on the interactions among the capsid protein, the newly identified scaffolding proteins, and the portal complex. A similar viral mechanism may be found in some dsDNA viruses.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
