

PIKE-mediated PI3-kinase activity is required for AMPA receptor surface expression
- PMID: 21847098
- PMCID: PMC3199380
- DOI: 10.1038/emboj.2011.281
PIKE-mediated PI3-kinase activity is required for AMPA receptor surface expression
Abstract
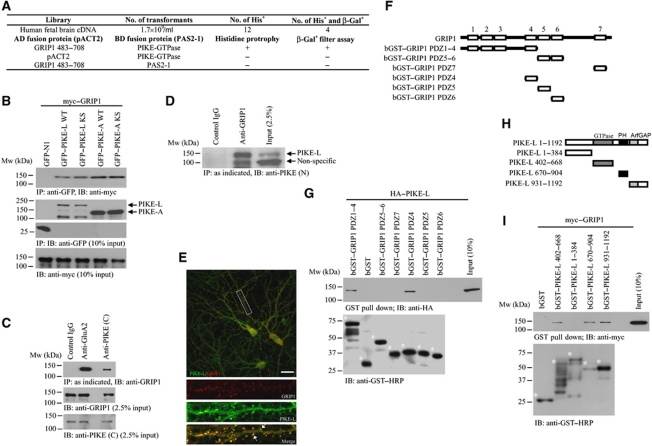
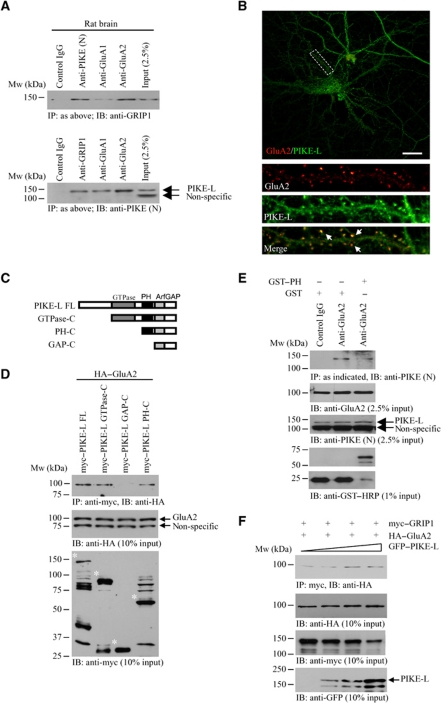
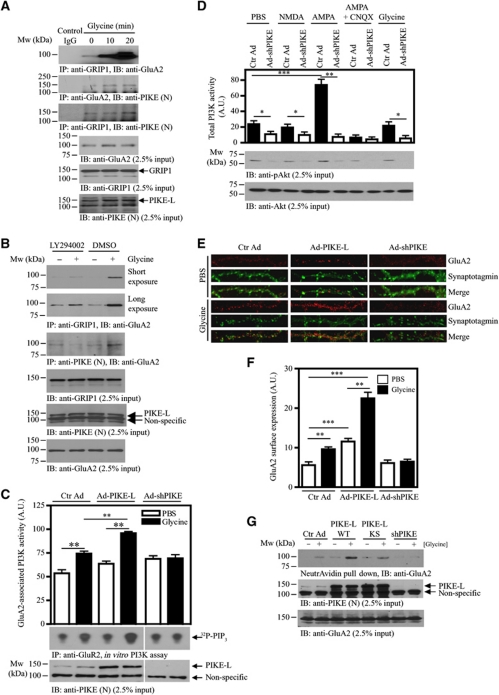
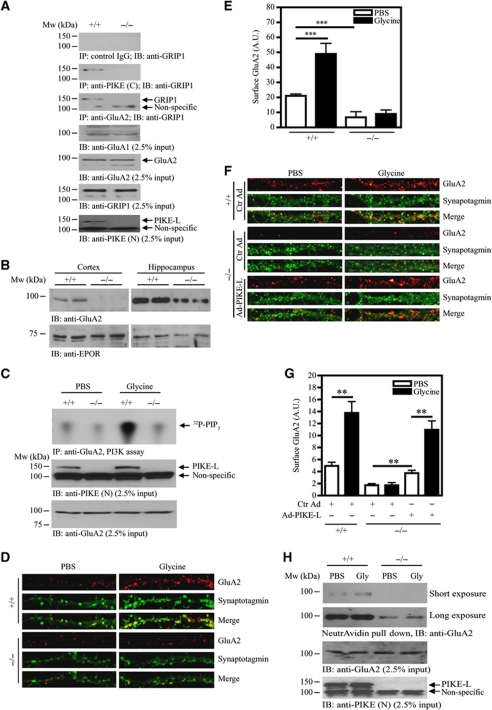
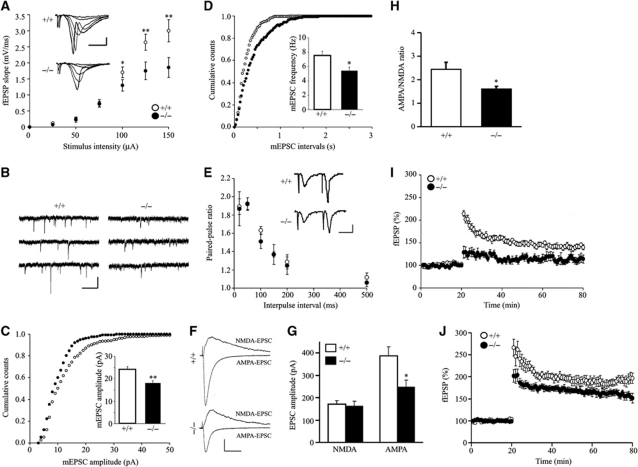
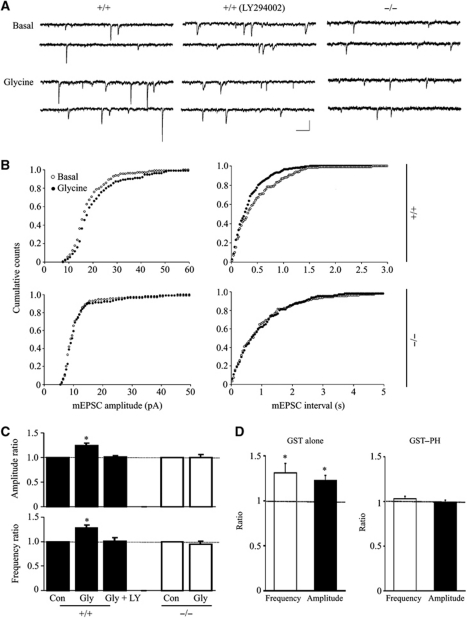
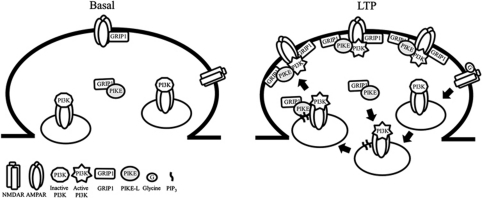
AMPAR (α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid receptor) is an ion channel involved in the formation of synaptic plasticity. However, the molecular mechanism that couples plasticity stimuli to the trafficking of postsynaptic AMPAR remains poorly understood. Here, we show that PIKE (phosphoinositide 3-kinase enhancer) GTPases regulate neuronal AMPAR activity by promoting GluA2/GRIP1 association. PIKE-L directly interacts with both GluA2 and GRIP1 and forms a tertiary complex upon glycine-induced NMDA receptor activation. PIKE-L is also essential for glycine-induced GluA2-associated PI3K activation. Genetic ablation of PIKE (PIKE(-/-)) in neurons suppresses GluA2-associated PI3K activation, therefore inhibiting the subsequent surface expression of GluA2 and the formation of long-term potentiation. Our findings suggest that PIKE-L is a critical factor in controlling synaptic AMPAR insertion.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
GRIP1 regulates synaptic plasticity and learning and memory.Proc Natl Acad Sci U S A. 2020 Oct 6;117(40):25085-25091. doi: 10.1073/pnas.2014827117. Epub 2020 Sep 18. Proc Natl Acad Sci U S A. 2020. PMID: 32948689 Free PMC article.
-
Acute BDNF treatment upregulates GluR1-SAP97 and GluR2-GRIP1 interactions: implications for sustained AMPA receptor expression.PLoS One. 2013;8(2):e57124. doi: 10.1371/journal.pone.0057124. Epub 2013 Feb 27. PLoS One. 2013. PMID: 23460828 Free PMC article.
-
Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1N.Cell. 2000 Dec 8;103(6):919-30. doi: 10.1016/s0092-8674(00)00195-1. Cell. 2000. PMID: 11136977
-
Regulation of neuronal PKA signaling through AKAP targeting dynamics.Eur J Cell Biol. 2006 Jul;85(7):627-33. doi: 10.1016/j.ejcb.2006.01.010. Epub 2006 Feb 28. Eur J Cell Biol. 2006. PMID: 16504338 Review.
-
PIKE GTPase are phosphoinositide-3-kinase enhancers, suppressing programmed cell death.J Cell Mol Med. 2007 Jan-Feb;11(1):39-53. doi: 10.1111/j.1582-4934.2007.00014.x. J Cell Mol Med. 2007. PMID: 17367500 Free PMC article. Review.
Cited by
-
Coupling mechanical forces to electrical signaling: molecular motors and the intracellular transport of ion channels.Neuroscientist. 2013 Apr;19(2):145-59. doi: 10.1177/1073858412456088. Epub 2012 Aug 20. Neuroscientist. 2013. PMID: 22910031 Free PMC article. Review.
-
Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome.Cell Rep. 2015 May 5;11(5):727-36. doi: 10.1016/j.celrep.2015.03.060. Epub 2015 Apr 23. Cell Rep. 2015. PMID: 25921541 Free PMC article.
-
PIKE is essential for oligodendroglia development and CNS myelination.Proc Natl Acad Sci U S A. 2014 Feb 4;111(5):1993-8. doi: 10.1073/pnas.1318185111. Epub 2014 Jan 21. Proc Natl Acad Sci U S A. 2014. PMID: 24449917 Free PMC article.
-
The role of PI3K-mediated AMPA receptor changes in post-conditioning of propofol in brain protection.BMC Neurosci. 2019 Oct 1;20(1):51. doi: 10.1186/s12868-019-0532-6. BMC Neurosci. 2019. PMID: 31570094 Free PMC article.
-
Fasting and Systemic Insulin Signaling Regulate Phosphorylation of Brain Proteins That Modulate Cell Morphology and Link to Neurological Disorders.J Biol Chem. 2015 Dec 11;290(50):30030-41. doi: 10.1074/jbc.M115.668103. Epub 2015 Oct 23. J Biol Chem. 2015. PMID: 26499801 Free PMC article.
References
-
- Ahn JY, Rong R, Kroll TG, Van Meir EG, Snyder SH, Ye K (2004) PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J Biol Chem 279: 16441–16451 - PubMed
-
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39 - PubMed
-
- Burnashev N, Monyer H, Seeburg PH, Sakmann B (1992) Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8: 189–198 - PubMed
-
- Capocchi G, Zampolini M, Larson J (1992) Theta burst stimulation is optimal for induction of LTP at both apical and basal dendritic synapses on hippocampal CA1 neurons. Brain Res 591: 332–336 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
