

Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation
- PMID: 21852959
- PMCID: PMC3154961
- DOI: 10.1371/journal.pgen.1002228
Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation
Abstract
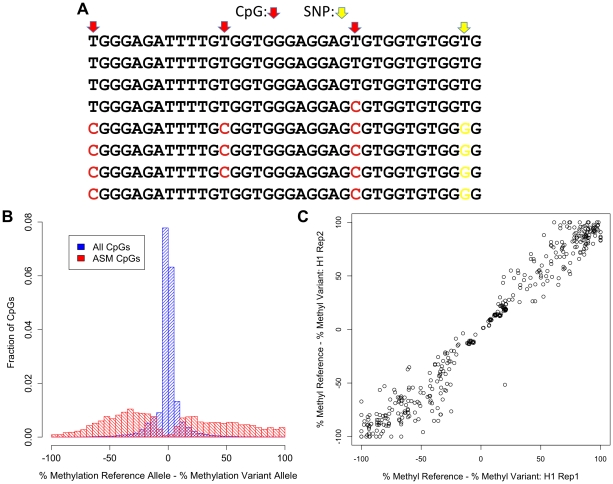
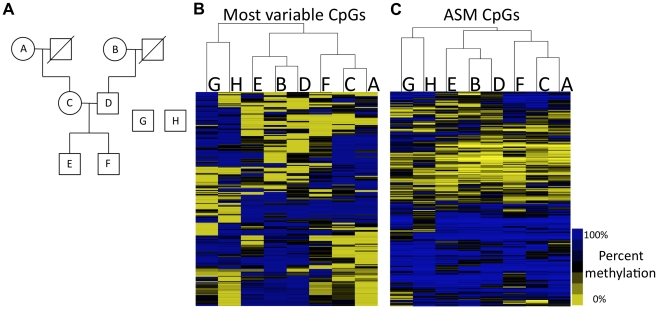
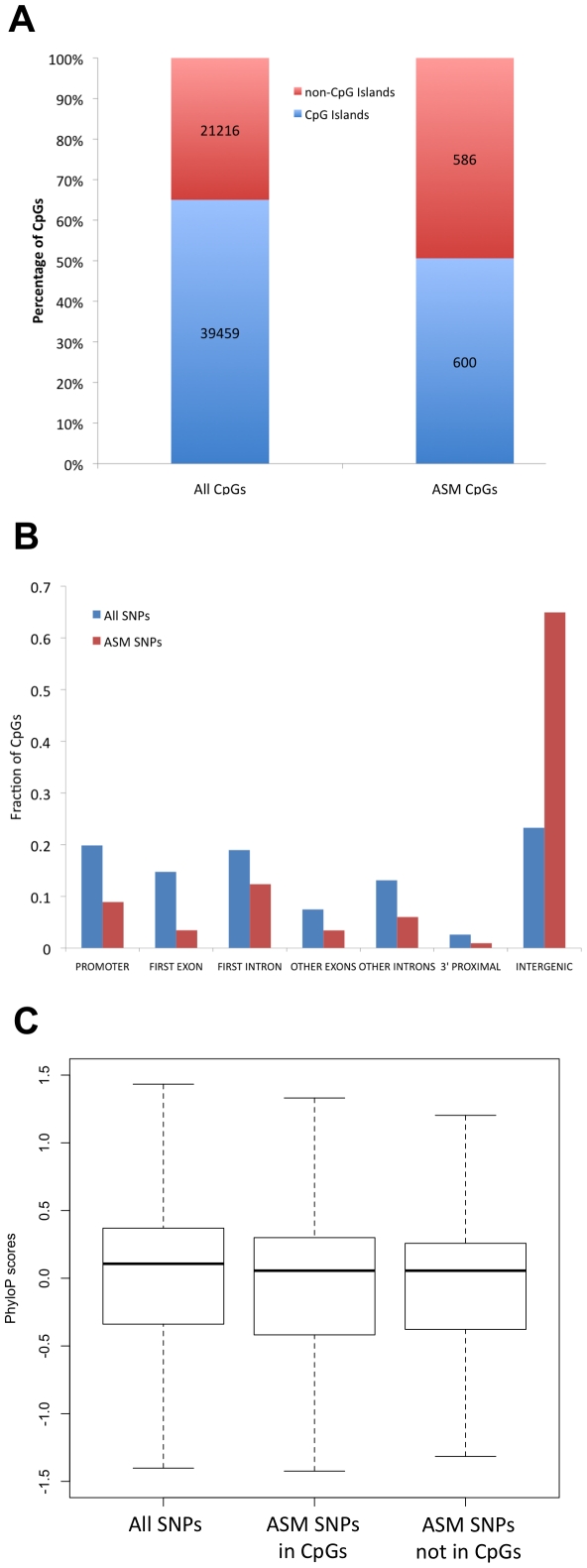
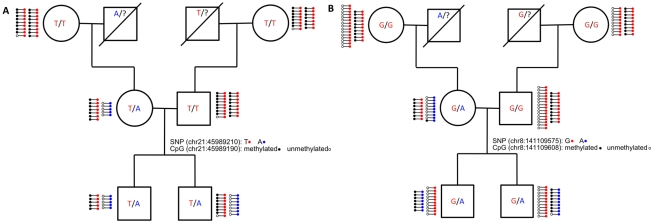
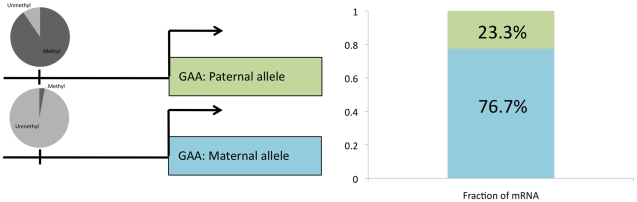
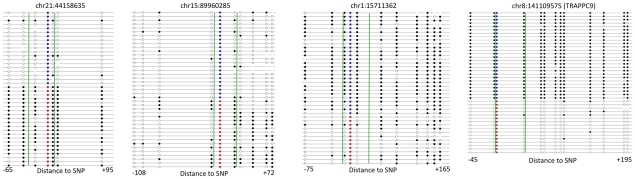
The methylation of cytosines in CpG dinucleotides is essential for cellular differentiation and the progression of many cancers, and it plays an important role in gametic imprinting. To assess variation and inheritance of genome-wide patterns of DNA methylation simultaneously in humans, we applied reduced representation bisulfite sequencing (RRBS) to somatic DNA from six members of a three-generation family. We observed that 8.1% of heterozygous SNPs are associated with differential methylation in cis, which provides a robust signature for Mendelian transmission and relatedness. The vast majority of differential methylation between homologous chromosomes (>92%) occurs on a particular haplotype as opposed to being associated with the gender of the parent of origin, indicating that genotype affects DNA methylation of far more loci than does gametic imprinting. We found that 75% of genotype-dependent differential methylation events in the family are also seen in unrelated individuals and that overall genotype can explain 80% of the variation in DNA methylation. These events are under-represented in CpG islands, enriched in intergenic regions, and located in regions of low evolutionary conservation. Even though they are generally not in functionally constrained regions, 22% (twice as many as expected by chance) of genes harboring genotype-dependent DNA methylation exhibited allele-specific gene expression as measured by RNA-seq of a lymphoblastoid cell line, indicating that some of these events are associated with gene expression differences. Overall, our results demonstrate that the influence of genotype on patterns of DNA methylation is widespread in the genome and greatly exceeds the influence of imprinting on genome-wide methylation patterns.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. - PubMed
-
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. - PubMed
-
- Jones PA. DNA methylation and cancer. Oncogene. 2002;21:5358–5360. - PubMed
-
- Richardson B. DNA methylation and autoimmune disease. Clin Immunol. 2003;109:72–79. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
