

Linking RNA polymerase backtracking to genome instability in E. coli
- PMID: 21854980
- PMCID: PMC3160732
- DOI: 10.1016/j.cell.2011.07.034
Linking RNA polymerase backtracking to genome instability in E. coli
Abstract
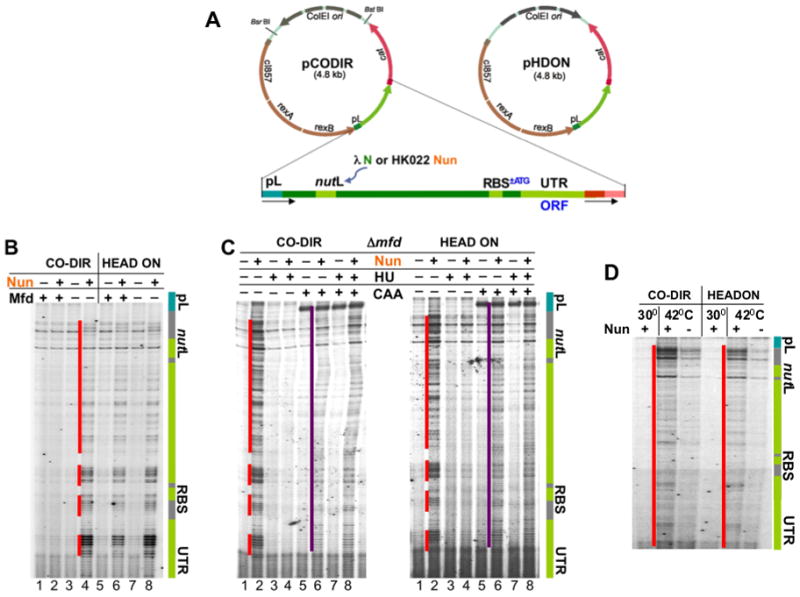
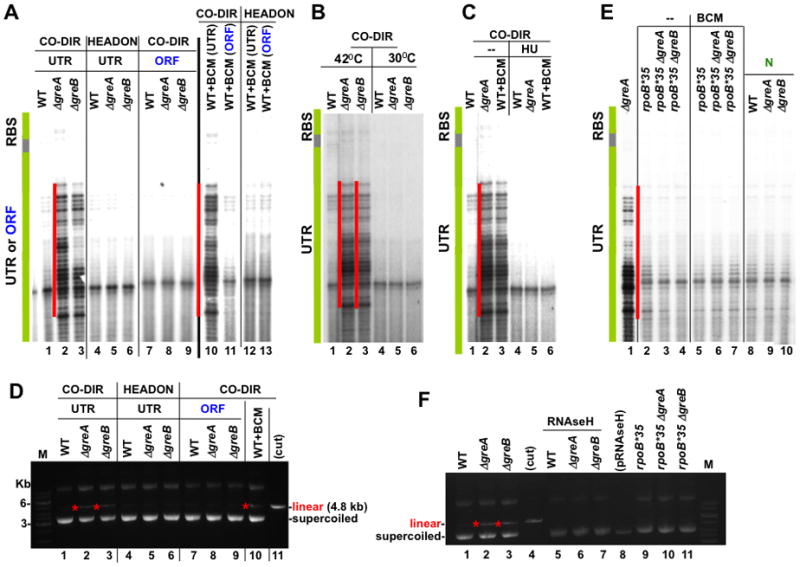
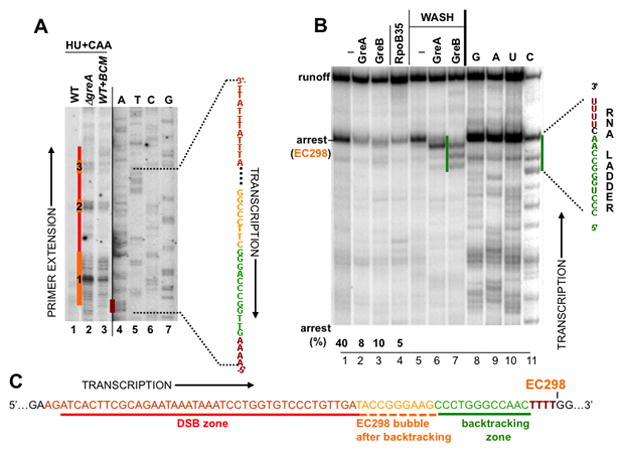
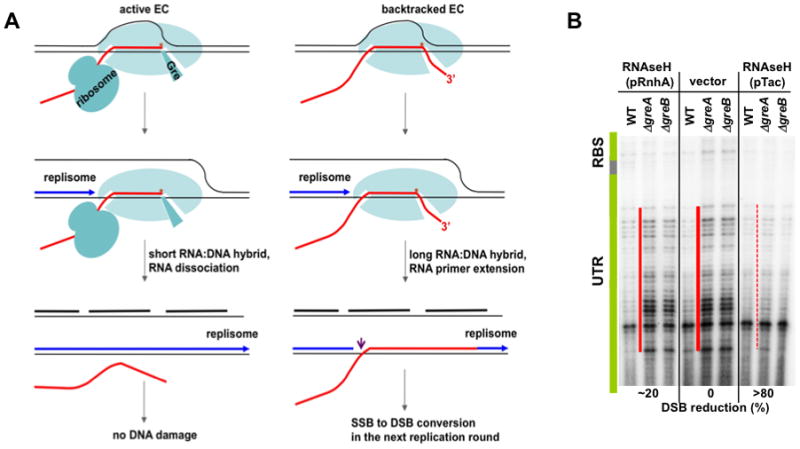
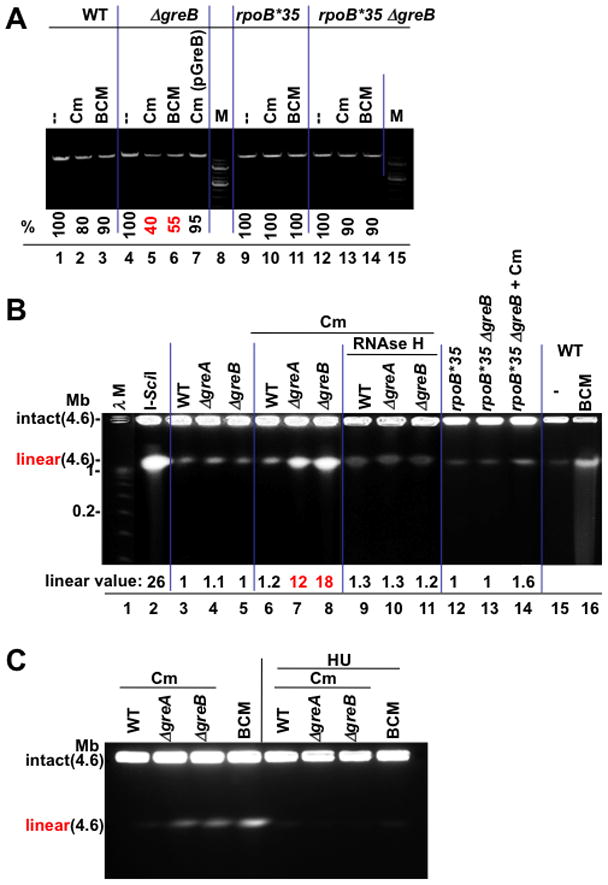
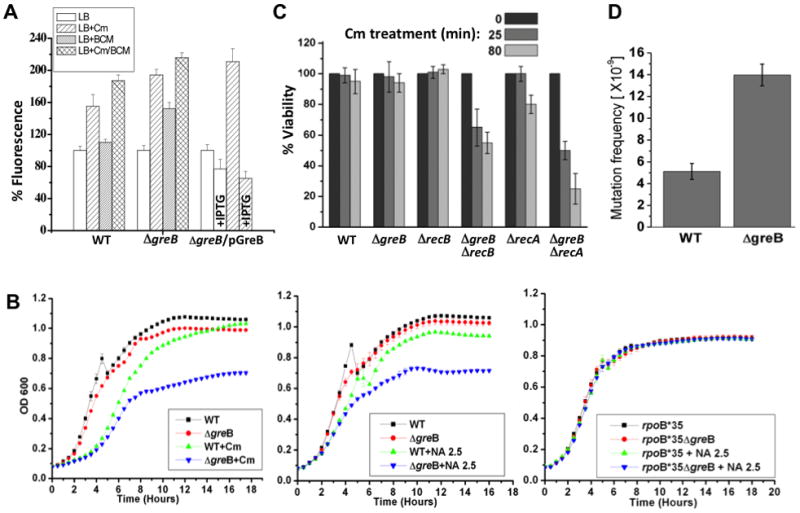
Frequent codirectional collisions between the replisome and RNA polymerase (RNAP) are inevitable because the rate of replication is much faster than that of transcription. Here we show that, in E. coli, the outcome of such collisions depends on the productive state of transcription elongation complexes (ECs). Codirectional collisions with backtracked (arrested) ECs lead to DNA double-strand breaks (DSBs), whereas head-on collisions do not. A mechanistic model is proposed to explain backtracking-mediated DSBs. We further show that bacteria employ various strategies to avoid replisome collisions with backtracked RNAP, the most general of which is translation that prevents RNAP backtracking. If translation is abrogated, DSBs are suppressed by elongation factors that either prevent backtracking or reactivate backtracked ECs. Finally, termination factors also contribute to genomic stability by removing arrested ECs. Our results establish RNAP backtracking as the intrinsic hazard to chromosomal integrity and implicate active ribosomes and other anti-backtracking mechanisms in genome maintenance.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Alff-Steinberger C. Comparative study of mutations in Escherichia coli and Salmonella typhimurium shows that codon conservation is strongly correlated with codon usage. J Theor Biol. 2000;206:307–311. - PubMed
-
- Birren B, Lai E. Pulse Field Gel Electrophoresis, a Practical Guide. Academic Press; New York: 1993.
-
- Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. - PubMed
-
- Borukhov S, Lee J, Laptenko O. Bacterial transcription elongation factors: new insights into molecular mechanism of action. Mol Microbiol. 2005;55:1315–1324. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
