

Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia
- PMID: 21857683
- PMCID: PMC3169705
- DOI: 10.1038/nature10353
Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia
Abstract
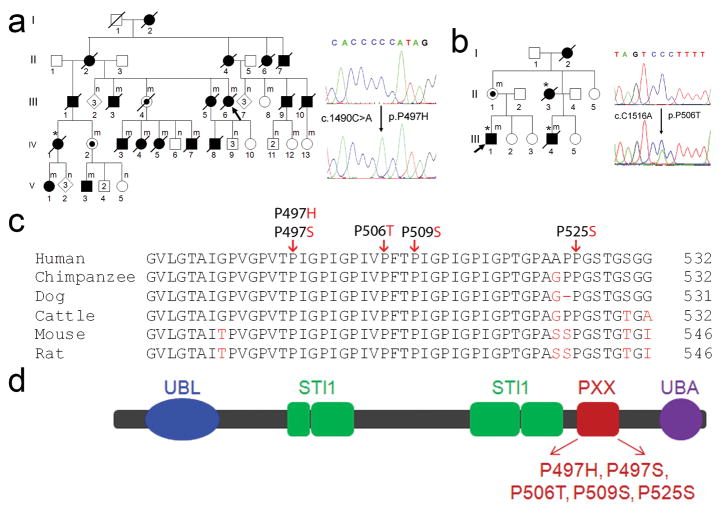
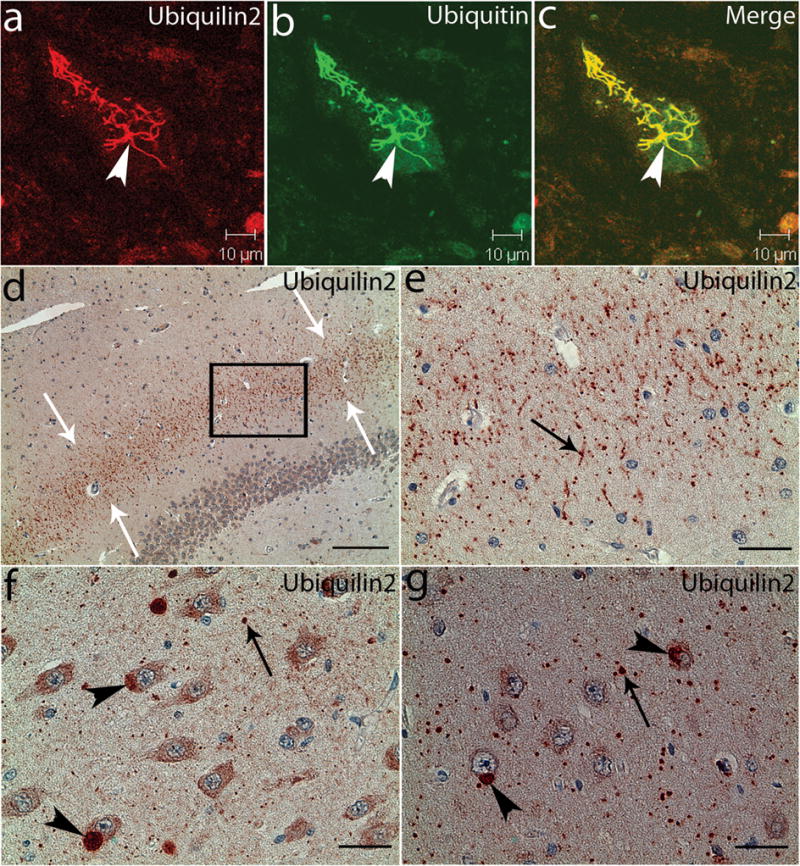
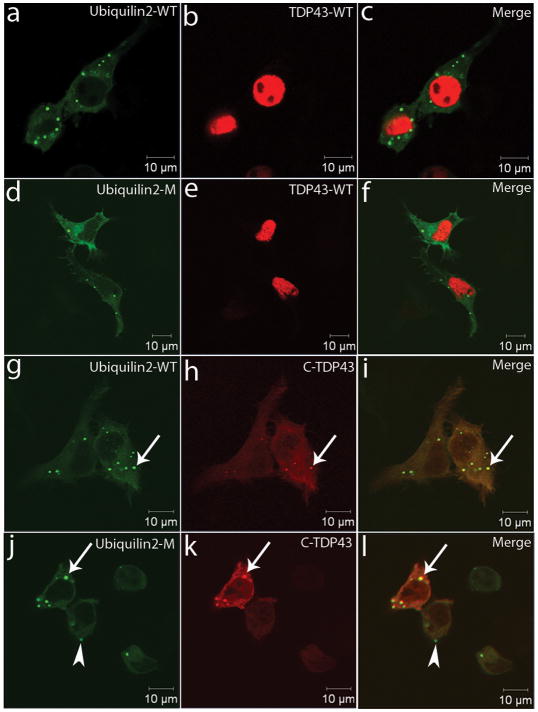
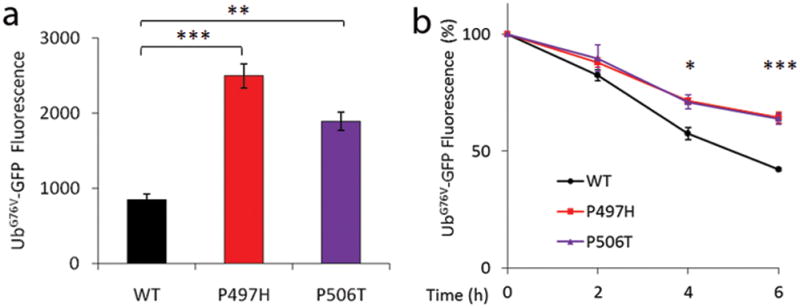
Amyotrophic lateral sclerosis (ALS) is a paralytic and usually fatal disorder caused by motor-neuron degeneration in the brain and spinal cord. Most cases of ALS are sporadic but about 5-10% are familial. Mutations in superoxide dismutase 1 (SOD1), TAR DNA-binding protein (TARDBP, also known as TDP43) and fused in sarcoma (FUS, also known as translocated in liposarcoma (TLS)) account for approximately 30% of classic familial ALS. Mutations in several other genes have also been reported as rare causes of ALS or ALS-like syndromes. The causes of the remaining cases of familial ALS and of the vast majority of sporadic ALS are unknown. Despite extensive studies of previously identified ALS-causing genes, the pathogenic mechanism underlying motor-neuron degeneration in ALS remains largely obscure. Dementia, usually of the frontotemporal lobar type, may occur in some ALS cases. It is unclear whether ALS and dementia share common aetiology and pathogenesis in ALS/dementia. Here we show that mutations in UBQLN2, which encodes the ubiquitin-like protein ubiquilin 2, cause dominantly inherited, chromosome-X-linked ALS and ALS/dementia. We describe novel ubiquilin 2 pathology in the spinal cords of ALS cases and in the brains of ALS/dementia cases with or without UBQLN2 mutations. Ubiquilin 2 is a member of the ubiquilin family, which regulates the degradation of ubiquitinated proteins. Functional analysis showed that mutations in UBQLN2 lead to an impairment of protein degradation. Therefore, our findings link abnormalities in ubiquilin 2 to defects in the protein degradation pathway, abnormal protein aggregation and neurodegeneration, indicating a common pathogenic mechanism that can be exploited for therapeutic intervention.
Conflict of interest statement
Figures
Comment in
-
A role for ubiquilin 2 mutations in neurodegeneration.Nat Rev Neurol. 2011 Oct 11;7(11):599-600. doi: 10.1038/nrneurol.2011.163. Nat Rev Neurol. 2011. PMID: 21989241 No abstract available.
-
When 'UPS' fails to deliver: a novel gene associated with the ubiquitin-proteasome system causes familial ALS.Clin Genet. 2012 Feb;81(2):125. doi: 10.1111/j.1399-0004.2011.01815.x. Epub 2011 Dec 13. Clin Genet. 2012. PMID: 22092048 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
