

Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice
- PMID: 21859843
- PMCID: PMC3171098
- DOI: 10.1084/jem.20110571
Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice
Abstract
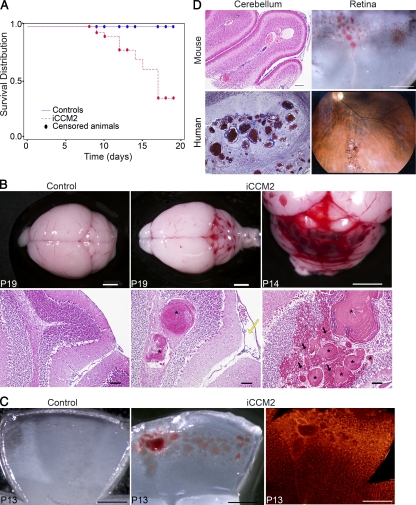
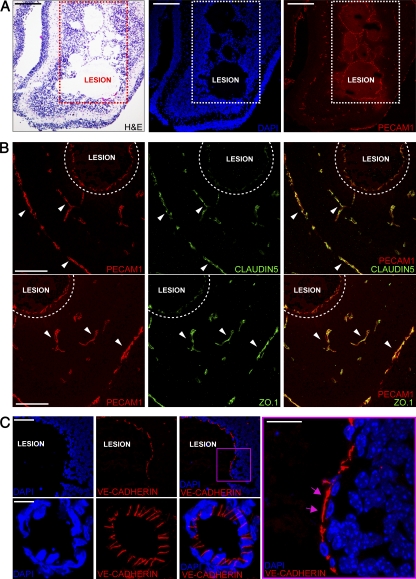
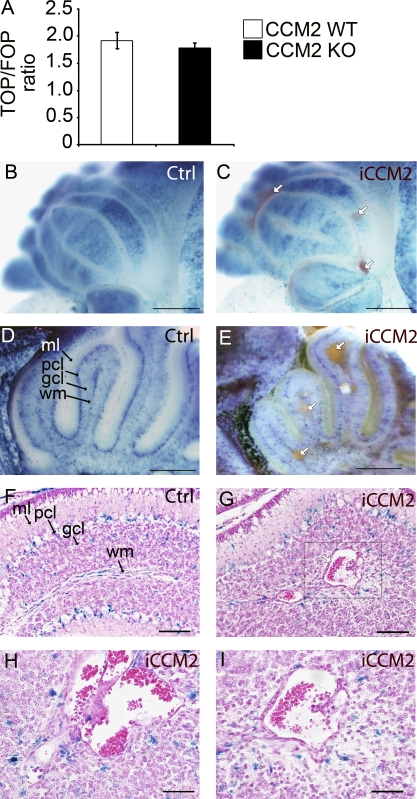
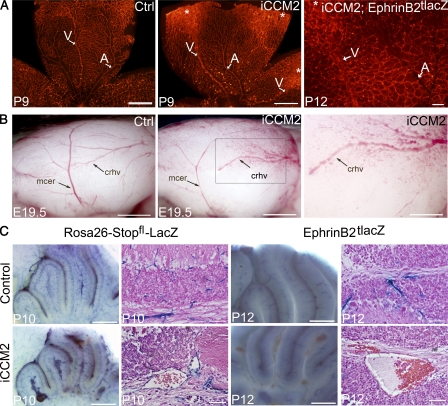
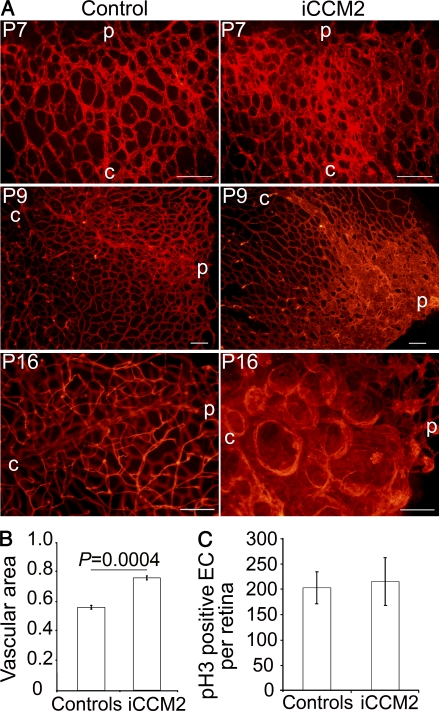
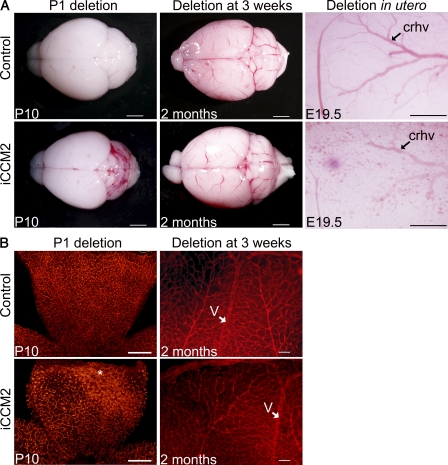
Cerebral cavernous malformations (CCM) are vascular malformations of the central nervous system (CNS) that lead to cerebral hemorrhages. Familial CCM occurs as an autosomal dominant condition caused by loss-of-function mutations in one of the three CCM genes. Constitutive or tissue-specific ablation of any of the Ccm genes in mice previously established the crucial role of Ccm gene expression in endothelial cells for proper angiogenesis. However, embryonic lethality precluded the development of relevant CCM mouse models. Here, we show that endothelial-specific Ccm2 deletion at postnatal day 1 (P1) in mice results in vascular lesions mimicking human CCM lesions. Consistent with CCM1/3 involvement in the same human disease, deletion of Ccm1/3 at P1 in mice results in similar CCM lesions. The lesions are located in the cerebellum and the retina, two organs undergoing intense postnatal angiogenesis. Despite a pan-endothelial Ccm2 deletion, CCM lesions are restricted to the venous bed. Notably, the consequences of Ccm2 loss depend on the developmental timing of Ccm2 ablation. This work provides a highly penetrant and relevant CCM mouse model.
© 2011 Boulday et al.
Figures
References
-
- Bergametti F., Denier C., Labauge P., Arnoult M., Boetto S., Clanet M., Coubes P., Echenne B., Ibrahim R., Irthum B., et al. ; SociétéFrançaise de Neurochirurgie 2005. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 76:42–51 10.1086/426952 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
