

Molecular control of endothelial cell behaviour during blood vessel morphogenesis
- PMID: 21860391
- PMCID: PMC3319719
- DOI: 10.1038/nrm3176
Molecular control of endothelial cell behaviour during blood vessel morphogenesis
Abstract
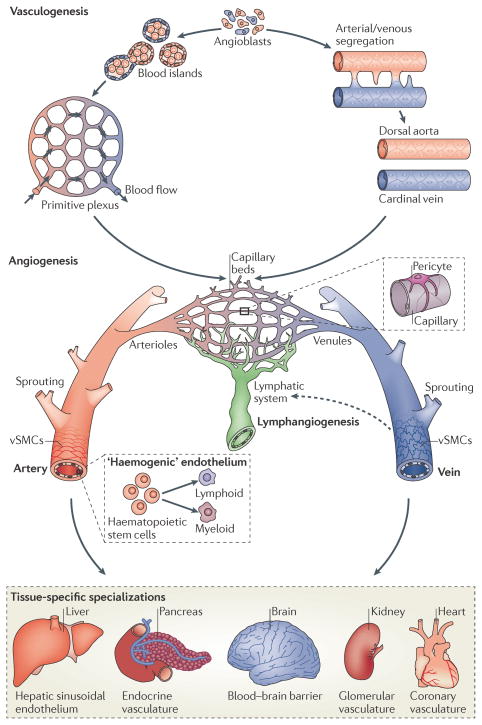
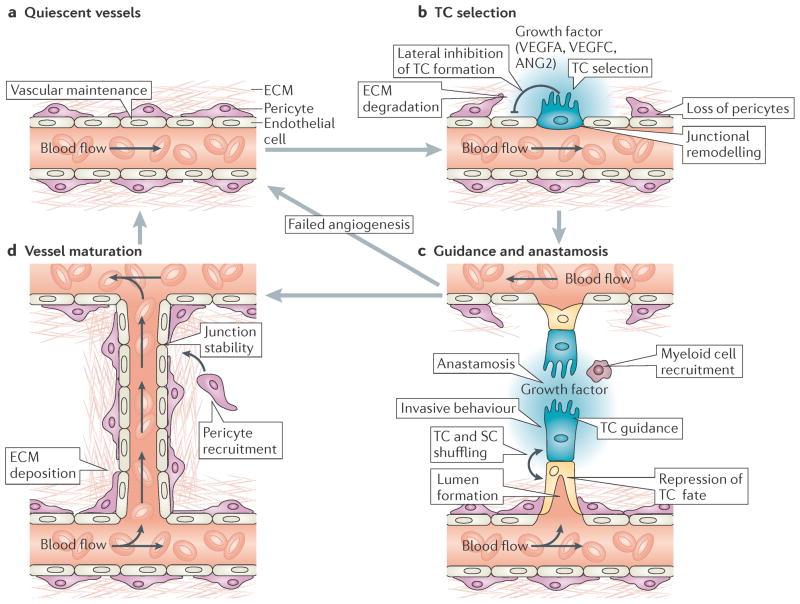
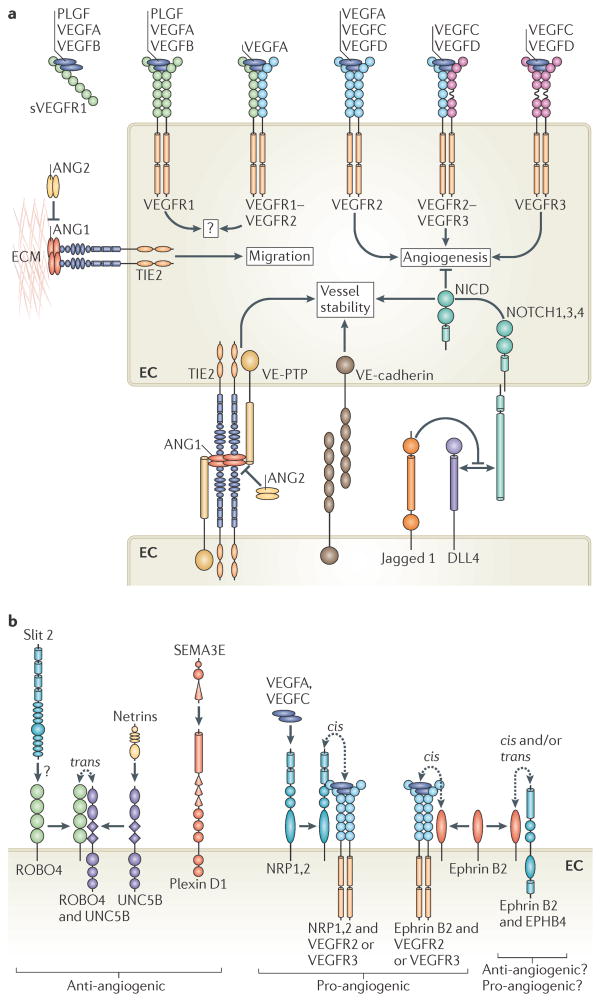
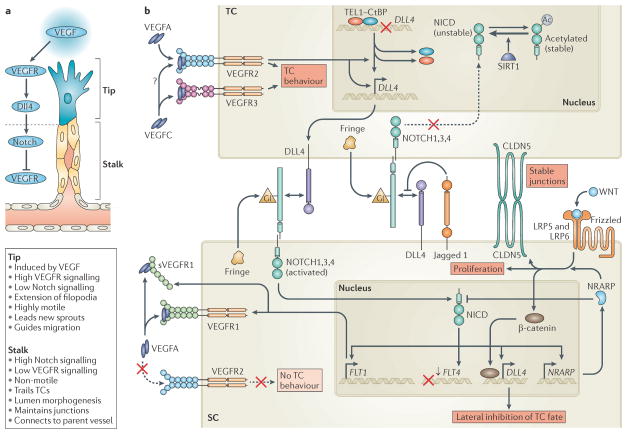
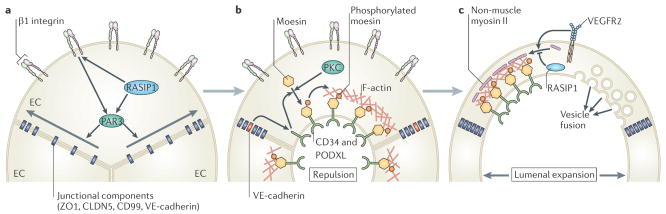
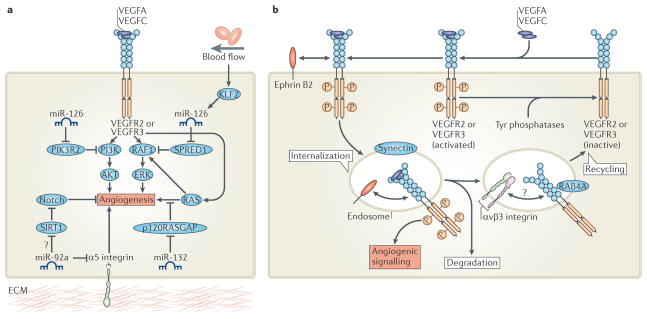
The vertebrate vasculature forms an extensive branched network of blood vessels that supplies tissues with nutrients and oxygen. During vascular development, coordinated control of endothelial cell behaviour at the levels of cell migration, proliferation, polarity, differentiation and cell-cell communication is critical for functional blood vessel morphogenesis. Recent data uncover elaborate transcriptional, post-transcriptional and post-translational mechanisms that fine-tune key signalling pathways (such as the vascular endothelial growth factor and Notch pathways) to control endothelial cell behaviour during blood vessel sprouting (angiogenesis). These emerging frameworks controlling angiogenesis provide unique insights into fundamental biological processes common to other systems, such as tissue branching morphogenesis, mechanotransduction and tubulogenesis.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nature Rev Mol Cell Biol. 2007;8:464–478. - PubMed
-
- Rocha SF, Adams RH. Molecular differentiation and specialization of vascular beds. Angiogenesis. 2009;12:139–147. - PubMed
-
- Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–476. - PubMed
-
- Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
