

Auto-activation of c-JUN gene by amino acid deprivation of hepatocellular carcinoma cells reveals a novel c-JUN-mediated signaling pathway
- PMID: 21862593
- PMCID: PMC3196148
- DOI: 10.1074/jbc.M111.277673
Auto-activation of c-JUN gene by amino acid deprivation of hepatocellular carcinoma cells reveals a novel c-JUN-mediated signaling pathway
Abstract
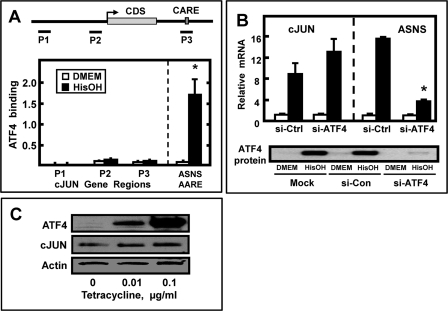
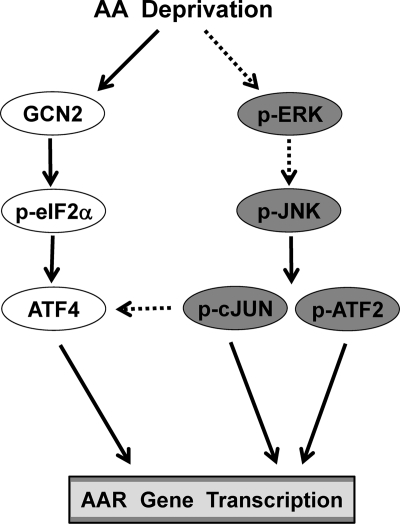
Mammalian cells respond to protein or amino acid (AA) limitation by activating a number of signaling pathways, collectively referred to as the AA response (AAR), that modulate a range of cellular functions, including transcriptional induction of target genes. This study demonstrates that in hepatocellular carcinoma cells, expression of c-JUN, JUN-B, c-FOS, and FOS-B was induced by the AAR, whereas JUN-D, FRA-1, and FRA-2 were not. Of the four activated FOS/JUN members, c-JUN made the largest contribution to the induction of several known AAR target genes. For several human liver, prostate, and ovarian cell lines, the AAR-induced increase in c-JUN expression was greater in transformed cells compared with nontransformed counterparts, an effect independent of cell growth rate. Thus far, the best characterized AA-responsive genes are all transcriptionally activated by ATF4, but the AAR-dependent induction of c-JUN transcription was ATF4-independent. The increased expression of c-JUN was dependent on ATF2 and on activation of the MEK-ERK and JNK arms of the MAPK signaling pathways. Formation of c-JUN-ATF2-activated heterodimers was increased after AA limitation, and c-JUN or ATF2 knockdown suppressed the induction of c-JUN and other AAR target genes. AA deprivation triggers a feed-forward process that involves phosphorylation of existing c-JUN protein by JNK and subsequent auto-activation of the c-JUN gene by recruitment of c-JUN and ATF2 to two AP-1 sites within the proximal promoter. The results document the novel observation that AP-1 sequences within the c-JUN gene can function as transcriptional amino acid-response elements.
Figures







Similar articles
-
MAPK signaling triggers transcriptional induction of cFOS during amino acid limitation of HepG2 cells.Biochim Biophys Acta. 2015 Mar;1853(3):539-48. doi: 10.1016/j.bbamcr.2014.12.013. Epub 2014 Dec 16. Biochim Biophys Acta. 2015. PMID: 25523140 Free PMC article.
-
Regulation of the ATF3 gene by a single promoter in response to amino acid availability and endoplasmic reticulum stress in human primary hepatocytes and hepatoma cells.Biochim Biophys Acta Gene Regul Mech. 2018 Feb;1861(2):72-79. doi: 10.1016/j.bbagrm.2018.01.002. Epub 2018 Feb 3. Biochim Biophys Acta Gene Regul Mech. 2018. PMID: 29413899 Free PMC article.
-
ERK signaling pathway is involved in p15INK4b/p16INK4a expression and HepG2 growth inhibition triggered by TPA and Saikosaponin a.Oncogene. 2003 Feb 20;22(7):955-63. doi: 10.1038/sj.onc.1206237. Oncogene. 2003. PMID: 12592382
-
The roles of ATF2 (activating transcription factor 2) in tumorigenesis.Biochem Soc Trans. 2012 Feb;40(1):230-4. doi: 10.1042/BST20110630. Biochem Soc Trans. 2012. PMID: 22260696 Review.
-
ATF4-dependent transcription mediates signaling of amino acid limitation.Trends Endocrinol Metab. 2009 Nov;20(9):436-43. doi: 10.1016/j.tem.2009.05.008. Epub 2009 Sep 30. Trends Endocrinol Metab. 2009. PMID: 19800252 Free PMC article. Review.
Cited by
-
Regulation of Hepatic Metabolism and Cell Growth by the ATF/CREB Family of Transcription Factors.Diabetes. 2021 Mar;70(3):653-664. doi: 10.2337/dbi20-0006. Diabetes. 2021. PMID: 33608424 Free PMC article. Review.
-
Asparagine synthetase: regulation by cell stress and involvement in tumor biology.Am J Physiol Endocrinol Metab. 2013 Apr 15;304(8):E789-99. doi: 10.1152/ajpendo.00015.2013. Epub 2013 Feb 12. Am J Physiol Endocrinol Metab. 2013. PMID: 23403946 Free PMC article. Review.
-
Effects of lysine and methionine on mRNA expression of candidate transcription factors by primary bovine mammary epithelial cells.PLoS One. 2024 Dec 20;19(12):e0305440. doi: 10.1371/journal.pone.0305440. eCollection 2024. PLoS One. 2024. PMID: 39705261 Free PMC article.
-
Oncogenic K-Ras4B Dimerization Enhances Downstream Mitogen-activated Protein Kinase Signaling.J Mol Biol. 2020 Feb 14;432(4):1199-1215. doi: 10.1016/j.jmb.2020.01.002. Epub 2020 Jan 10. J Mol Biol. 2020. PMID: 31931009 Free PMC article.
-
A circRNA-miRNA-mRNA network identification for exploring underlying pathogenesis and therapy strategy of hepatocellular carcinoma.J Transl Med. 2018 Aug 9;16(1):220. doi: 10.1186/s12967-018-1593-5. J Transl Med. 2018. PMID: 30092792 Free PMC article.
References
-
- Lillycrop K. A., Phillips E. S., Jackson A. A., Hanson M. A., Burdge G. C. (2005) J. Nutr. 135, 1382–1386 - PubMed
-
- Morgane P. J., Austin-LaFrance R., Bronzino J., Tonkiss J., Díaz-Cintra S., Cintra L., Kemper T., Galler J. R. (1993) Neurosci. Biobehav. Rev. 17, 91–128 - PubMed
-
- Morley J. E. (2009) Curr. Opin. Clin. Nutr. Metab. Care 12, 607–610 - PubMed
-
- Pedrini M. T., Levey A. S., Lau J., Chalmers T. C., Wang P. H. (1996) Ann. Intern. Med. 124, 627–632 - PubMed
-
- Zimmerman J. A., Malloy V., Krajcik R., Orentreich N. (2003) Exp. Gerontol. 38, 47–52 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
