

Prevention of estradiol 17beta-D-glucuronide-induced canalicular transporter internalization by hormonal modulation of cAMP in rat hepatocytes
- PMID: 21865596
- PMCID: PMC3192868
- DOI: 10.1091/mbc.E11-01-0047
Prevention of estradiol 17beta-D-glucuronide-induced canalicular transporter internalization by hormonal modulation of cAMP in rat hepatocytes
Abstract
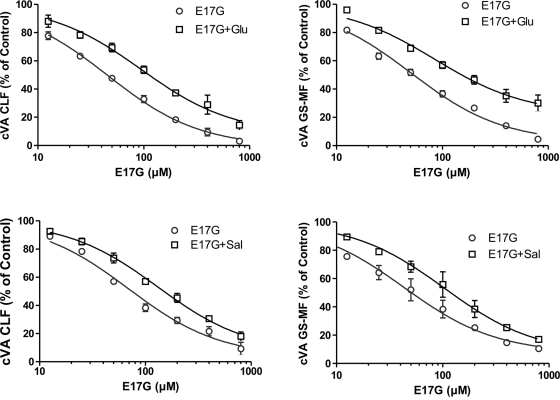
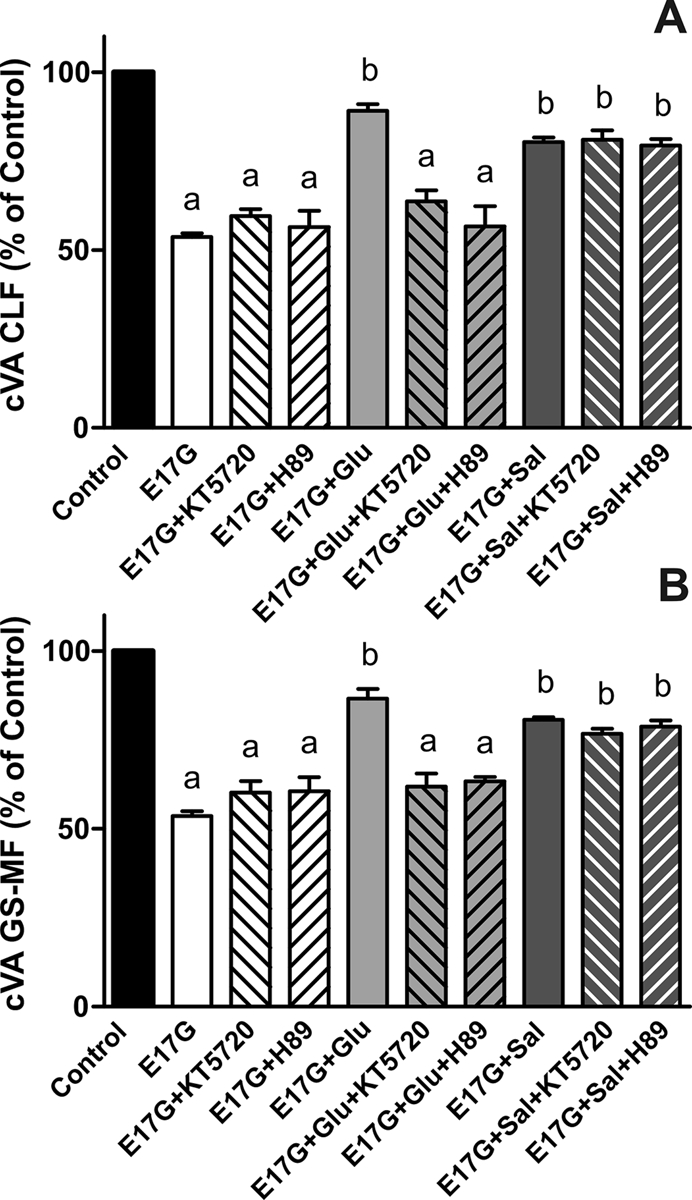
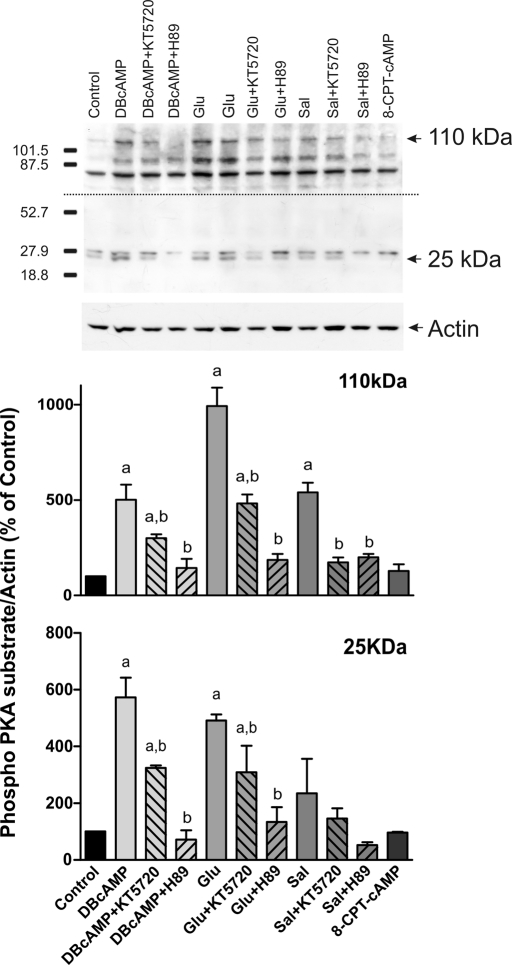
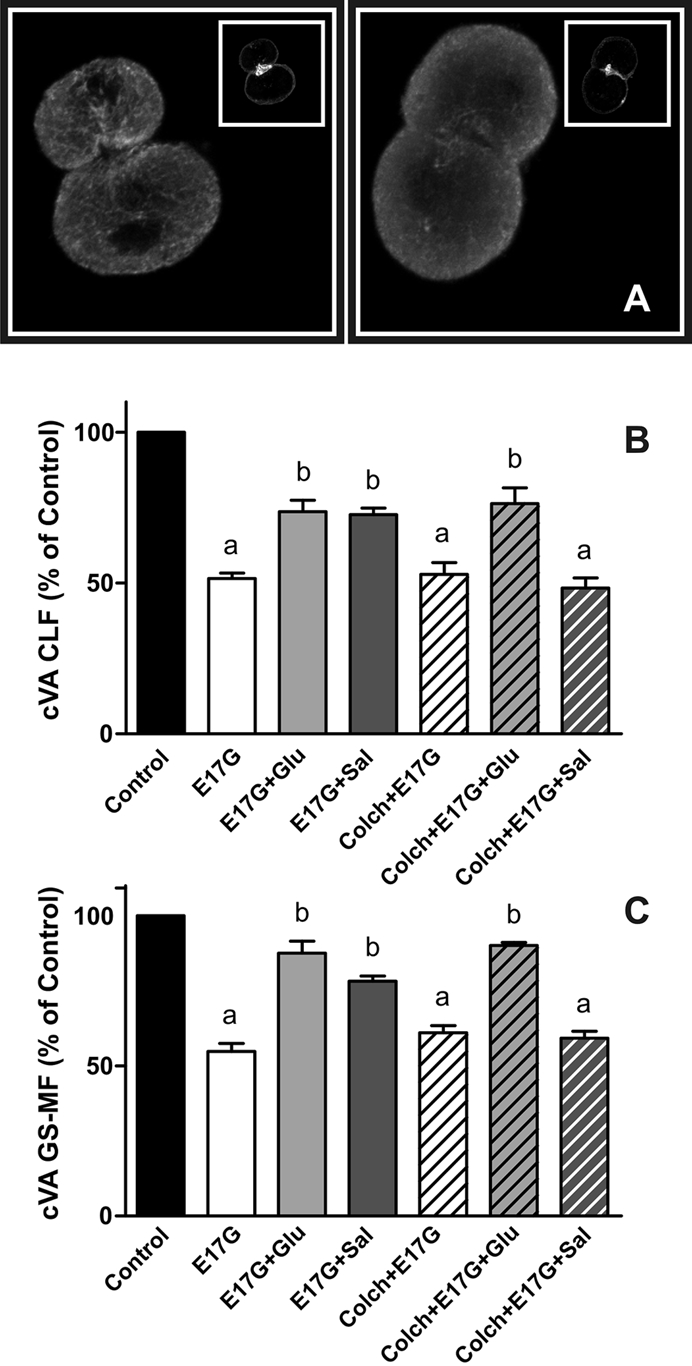
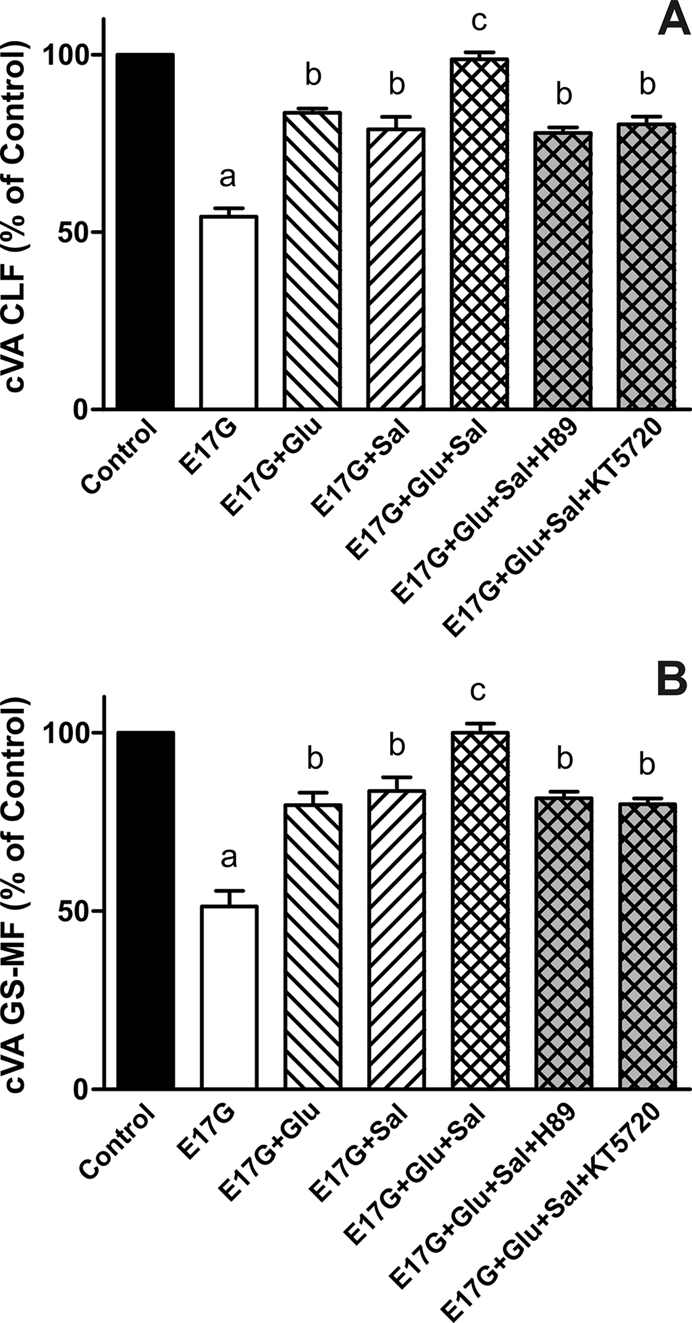
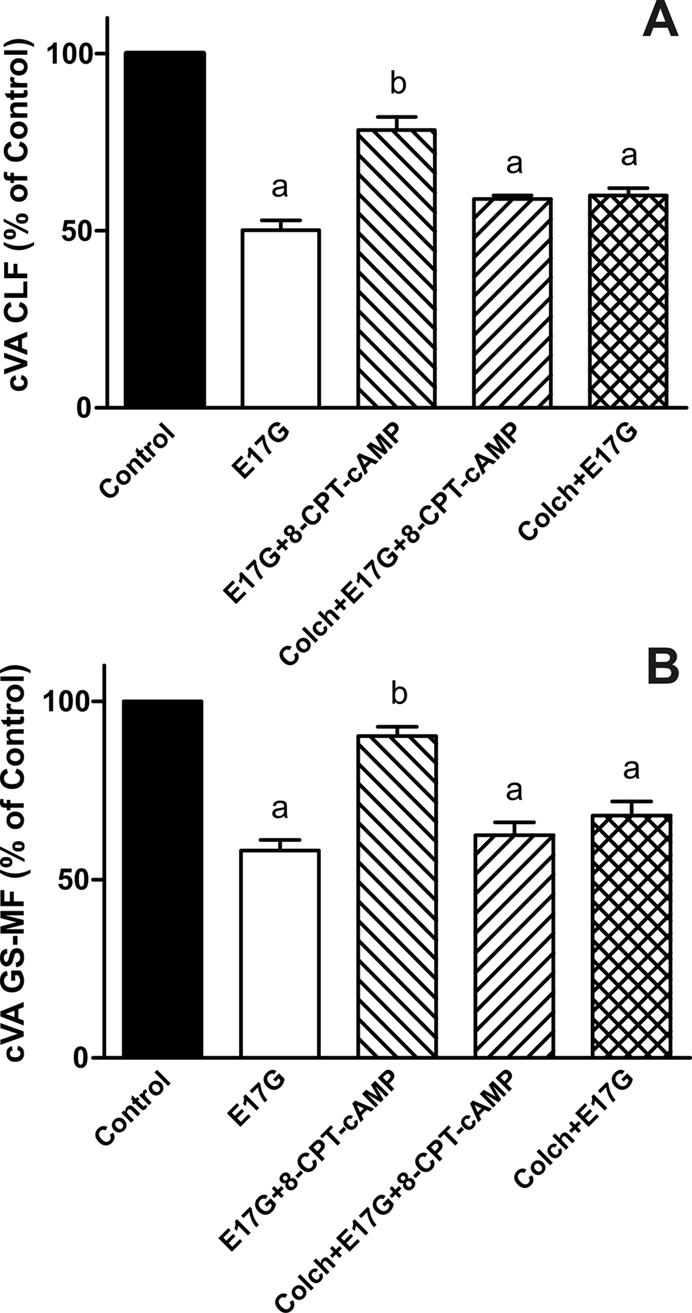
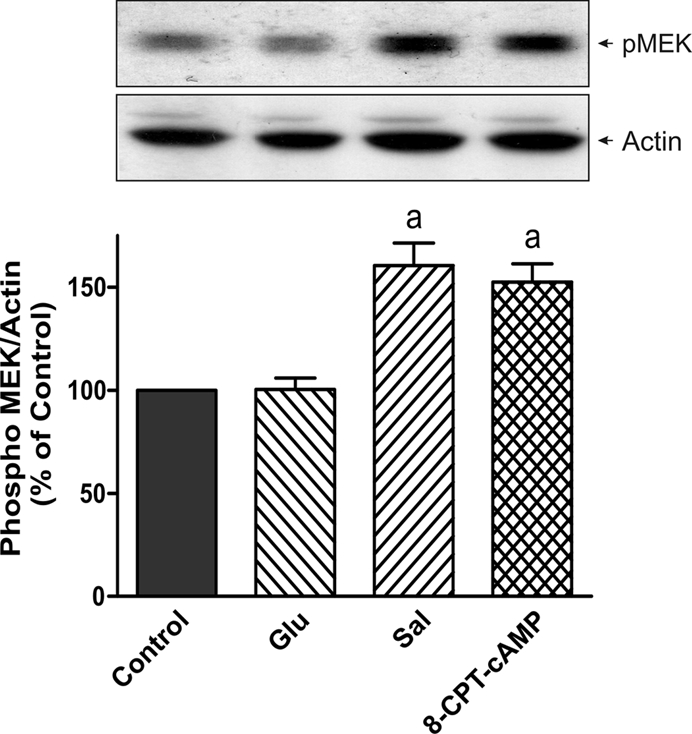
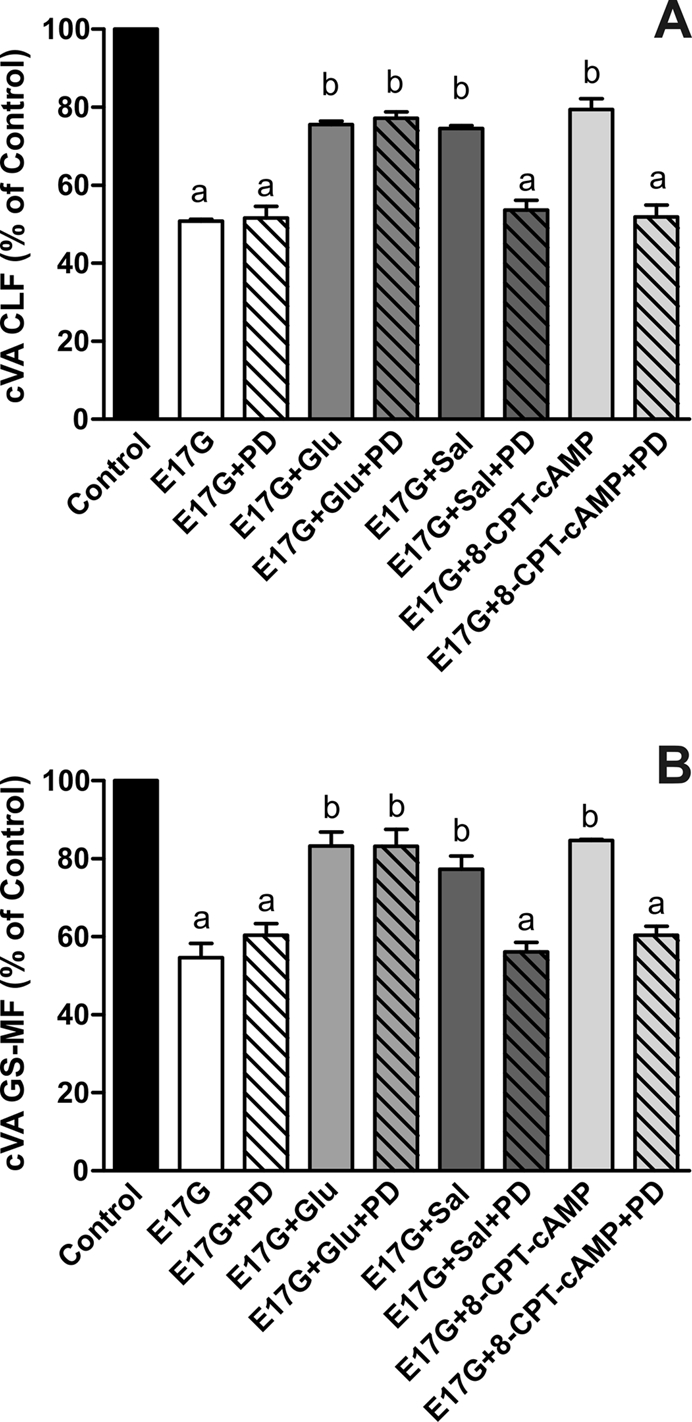
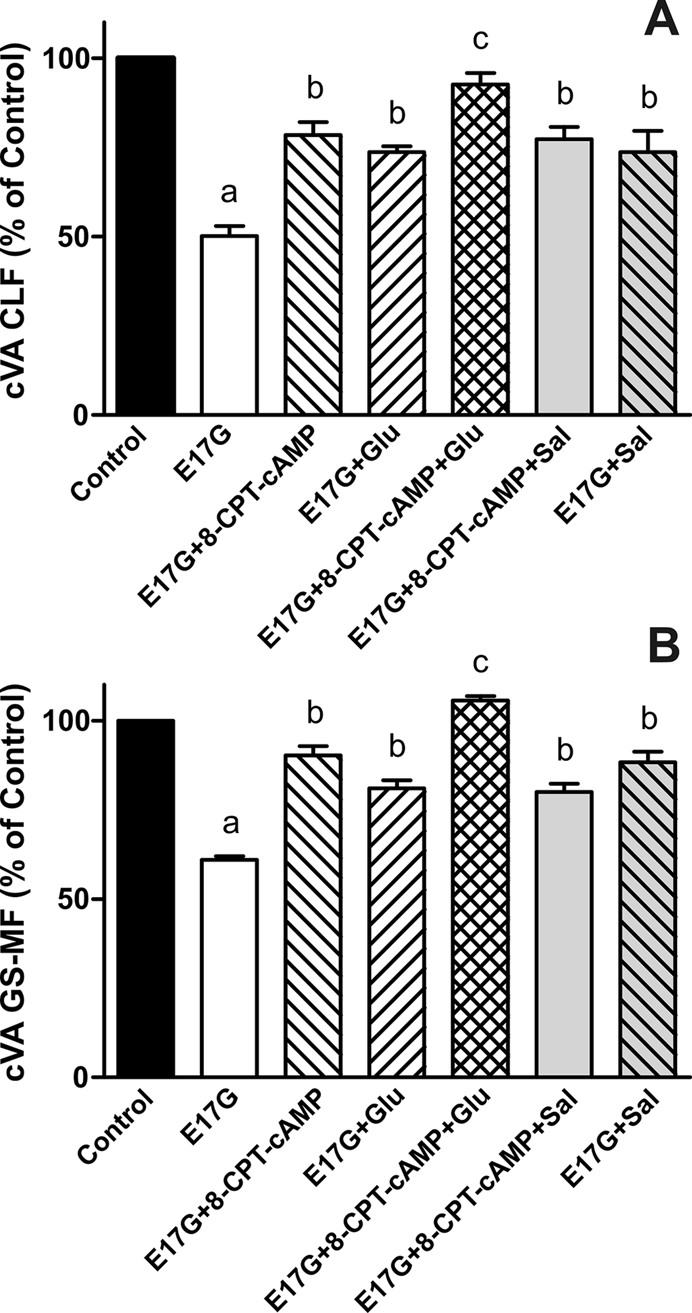
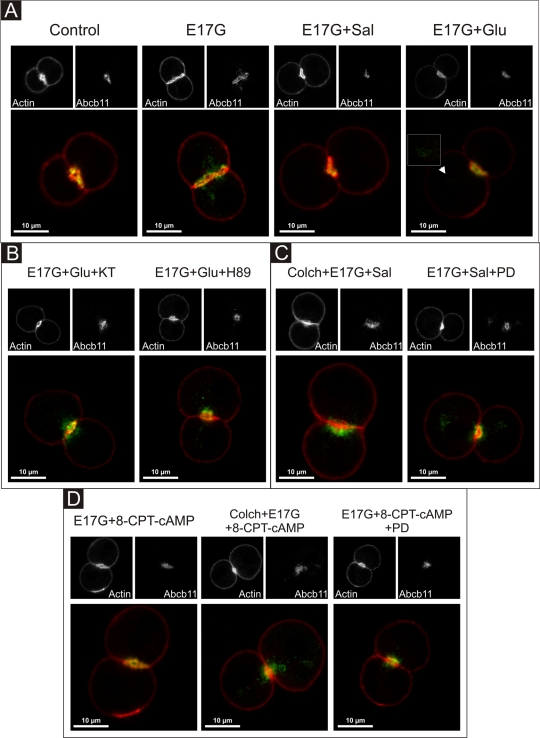
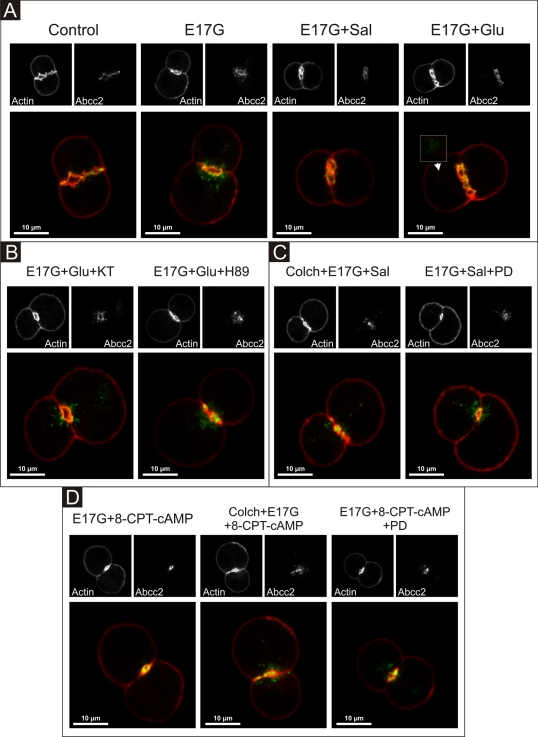
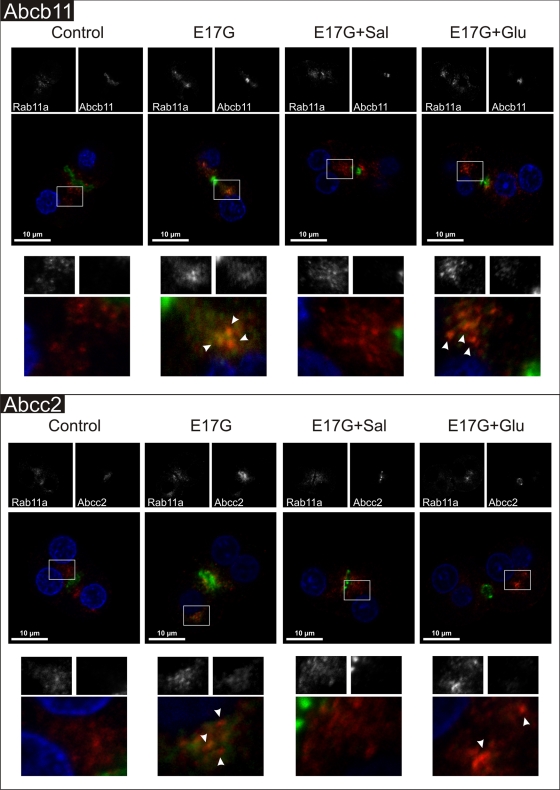
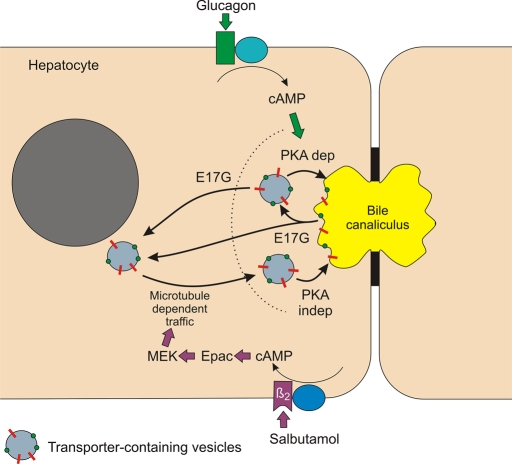
In estradiol 17β-d-glucuronide (E17G)-induced cholestasis, the canalicular hepatocellular transporters bile salt export pump (Abcb11) and multidrug-resistance associated protein 2 (Abcc2) undergo endocytic internalization. cAMP stimulates the trafficking of transporter-containing vesicles to the apical membrane and is able to prevent internalization of these transporters in estrogen-induced cholestasis. Hepatocyte levels of cAMP are regulated by hormones such as glucagon and adrenaline (via the β2 receptor). We analyzed the effects of glucagon and salbutamol (a β2 adrenergic agonist) on function and localization of Abcb11 and Abcc2 in isolated rat hepatocyte couplets exposed to E17G and compared the mechanistic bases of their effects. Glucagon and salbutamol partially prevented the impairment in Abcb11 and Abcc2 transport capacity. E17G also induced endocytic internalization of Abcb11 and Abcc2, which partially colocalized with the endosomal marker Rab11a. This effect was completely prevented by salbutamol, whereas some transporter-containing vesicles remained internalized and mainly colocalizing with Rab11a in the perinuclear region after incubation with glucagon. Glucagon prevention was dependent on cAMP-dependent protein kinase (PKA) and independent of exchange proteins activated directly by cAMP (Epac) and microtubules. In contrast, salbutamol prevention was PKA independent and Epac/MEK and microtubule dependent. Anticholestatic effects of glucagon and salbutamol were additive in nature. Our results show that increases in cAMP could activate different anticholestatic signaling pathways, depending on the hormonal mediator involved.
Figures
Similar articles
-
Hormonal modulation of hepatic cAMP prevents estradiol 17β-D-glucuronide-induced cholestasis in perfused rat liver.Dig Dis Sci. 2013 Jun;58(6):1602-14. doi: 10.1007/s10620-013-2558-4. Epub 2013 Jan 31. Dig Dis Sci. 2013. PMID: 23371010
-
G-protein-coupled receptor 30/adenylyl cyclase/protein kinase A pathway is involved in estradiol 17ß-D-glucuronide-induced cholestasis.Hepatology. 2014 Mar;59(3):1016-29. doi: 10.1002/hep.26752. Epub 2014 Jan 21. Hepatology. 2014. PMID: 24115158
-
Activation of insulin-like growth factor 1 receptor participates downstream of GPR30 in estradiol-17β-D-glucuronide-induced cholestasis in rats.Arch Toxicol. 2018 Feb;92(2):729-744. doi: 10.1007/s00204-017-2098-3. Epub 2017 Oct 31. Arch Toxicol. 2018. PMID: 29090346
-
Intracellular trafficking and regulation of canalicular ATP-binding cassette transporters.Semin Liver Dis. 2000;20(3):339-51. doi: 10.1055/s-2000-9388. Semin Liver Dis. 2000. PMID: 11076400 Review.
-
Regulation of synthesis and trafficking of canalicular transporters and its alteration in acquired hepatocellular cholestasis. Experimental therapeutic strategies for its prevention.Curr Med Chem. 2004 Feb;11(4):501-24. doi: 10.2174/0929867043455918. Curr Med Chem. 2004. PMID: 14965230 Review.
Cited by
-
Hormonal modulation of hepatic cAMP prevents estradiol 17β-D-glucuronide-induced cholestasis in perfused rat liver.Dig Dis Sci. 2013 Jun;58(6):1602-14. doi: 10.1007/s10620-013-2558-4. Epub 2013 Jan 31. Dig Dis Sci. 2013. PMID: 23371010
-
Carvedilol impairs bile acid homeostasis in mice: implication for nonalcoholic steatohepatitis.Toxicol Sci. 2023 Nov 28;196(2):200-217. doi: 10.1093/toxsci/kfad088. Toxicol Sci. 2023. PMID: 37632784 Free PMC article.
-
Targeted delivery of a phosphoinositide 3-kinase γ inhibitor to restore organ function in sepsis.EMBO Mol Med. 2021 Oct 7;13(10):e14436. doi: 10.15252/emmm.202114436. Epub 2021 Sep 2. EMBO Mol Med. 2021. PMID: 34472699 Free PMC article.
-
Mechanism of inhibition of taurolithocholate-induced retrieval of plasma membrane MRP2 by cyclic AMP and tauroursodeoxycholate.Physiol Rep. 2017 Dec;5(23):e13529. doi: 10.14814/phy2.13529. Physiol Rep. 2017. PMID: 29192063 Free PMC article.
-
Estradiol (E2) Improves Glucose-Stimulated Insulin Secretion and Stabilizes GDM Progression in a Prediabetic Mouse Model.Int J Mol Sci. 2022 Jun 15;23(12):6693. doi: 10.3390/ijms23126693. Int J Mol Sci. 2022. PMID: 35743136 Free PMC article.
References
-
- Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. - PubMed
-
- Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–377. - PubMed
-
- Bouscarel B, Matsuzaki Y, Le M, Gettys TW, Fromm H. Changes in G protein expression account for impaired modulation of hepatic cAMP formation after BDL. Am J Physiol. 1998;274:G1151–G1159. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
