

Combining markers into haplotypes can improve population structure inference
- PMID: 21868606
- PMCID: PMC3249356
- DOI: 10.1534/genetics.111.131136
Combining markers into haplotypes can improve population structure inference
Abstract
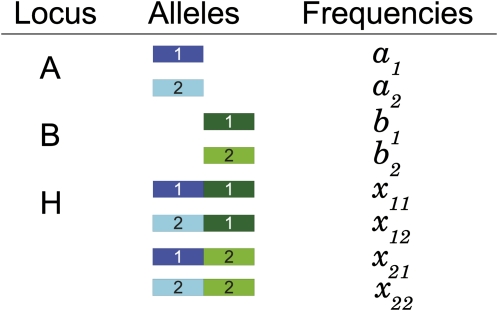
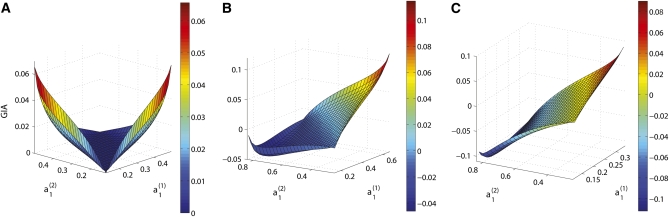
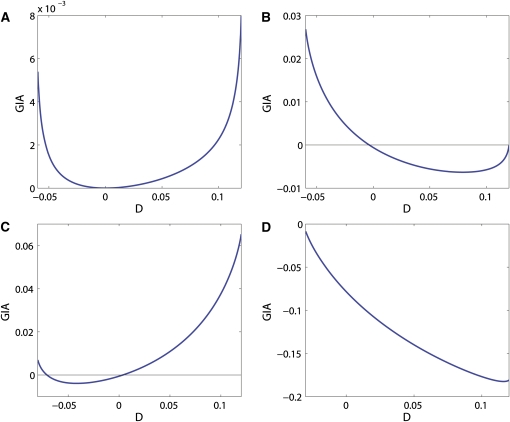
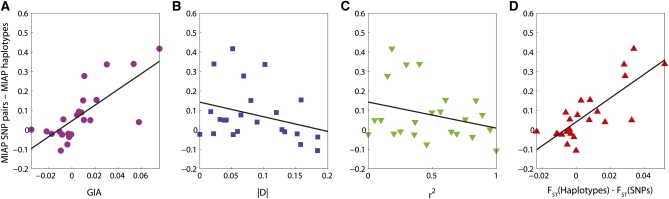
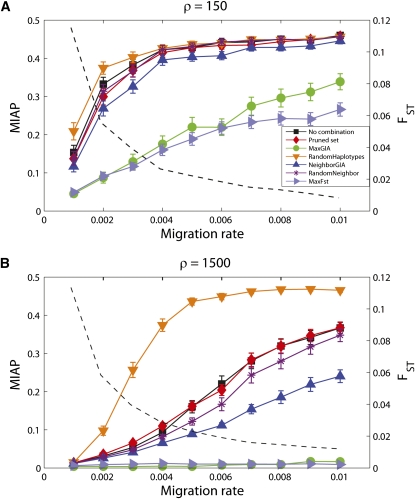
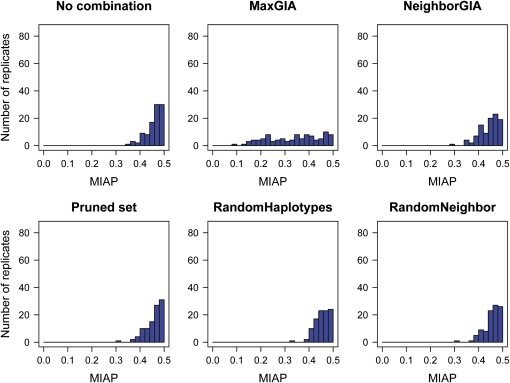
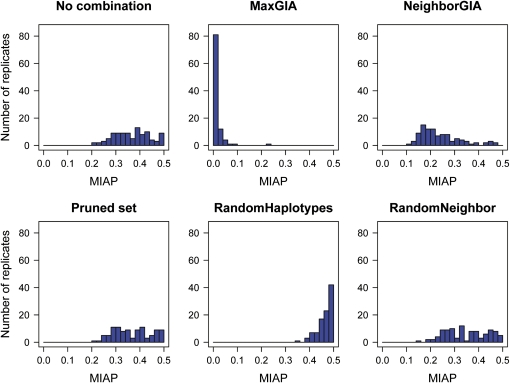
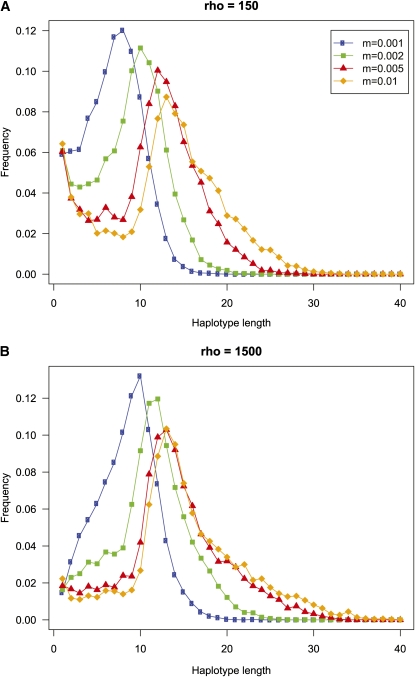
High-throughput genotyping and sequencing technologies can generate dense sets of genetic markers for large numbers of individuals. For most species, these data will contain many markers in linkage disequilibrium (LD). To utilize such data for population structure inference, we investigate the use of haplotypes constructed by combining the alleles at single-nucleotide polymorphisms (SNPs). We introduce a statistic derived from information theory, the gain of informativeness for assignment (GIA), which quantifies the additional information for assigning individuals to populations using haplotype data compared to using individual loci separately. Using a two-loci-two-allele model, we demonstrate that combining markers in linkage equilibrium into haplotypes always leads to nonpositive GIA, suggesting that combining the two markers is not advantageous for ancestry inference. However, for loci in LD, GIA is often positive, suggesting that assignment can be improved by combining markers into haplotypes. Using GIA as a criterion for combining markers into haplotypes, we demonstrate for simulated data a significant improvement of assigning individuals to candidate populations. For the many cases that we investigate, incorrect assignment was reduced between 26% and 97% using haplotype data. For empirical data from French and German individuals, the incorrectly assigned individuals can, for example, be decreased by 73% using haplotypes. Our results can be useful for challenging population structure and assignment problems, in particular for studies where large-scale population-genomic data are available.
Figures
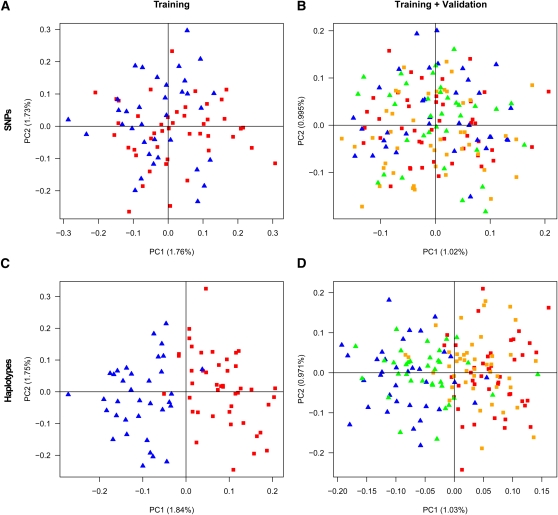
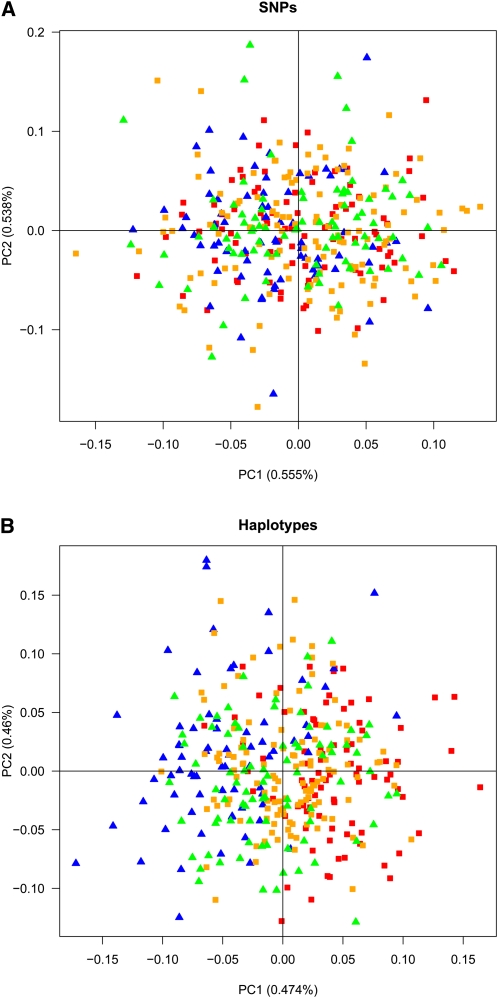
References
-
- Adams J. R., Lucash C., Schutte L., Waits L. P., 2007. Locating hybrid individuals in the red wolf (Canis rufus) experimental population area using a spatially targeted sampling strategy and faecal DNA genotyping. Mol. Ecol. 16: 1823–1834 - PubMed
-
- Aitken C. G. G., Taroni F., 2004. Statistics and the Evaluation of Evidence for Forensic Scientists, Ed. 2 John Wiley & Sons, New York
-
- Balding D. J., Nichols R. A., 1994. DNA profile match probability calculation: how to allow for population stratification, relatedness, database selection and single bands. Forensic Sci. Int. 64: 125–140 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
