

Inhibition of c-Jun-N-terminal kinase increases cardiac peroxisome proliferator-activated receptor alpha expression and fatty acid oxidation and prevents lipopolysaccharide-induced heart dysfunction
- PMID: 21873422
- PMCID: PMC3196095
- DOI: 10.1074/jbc.M111.272146
Inhibition of c-Jun-N-terminal kinase increases cardiac peroxisome proliferator-activated receptor alpha expression and fatty acid oxidation and prevents lipopolysaccharide-induced heart dysfunction
Abstract
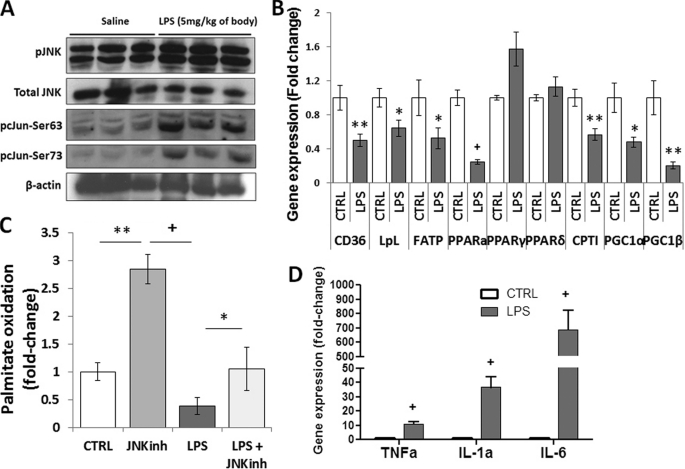
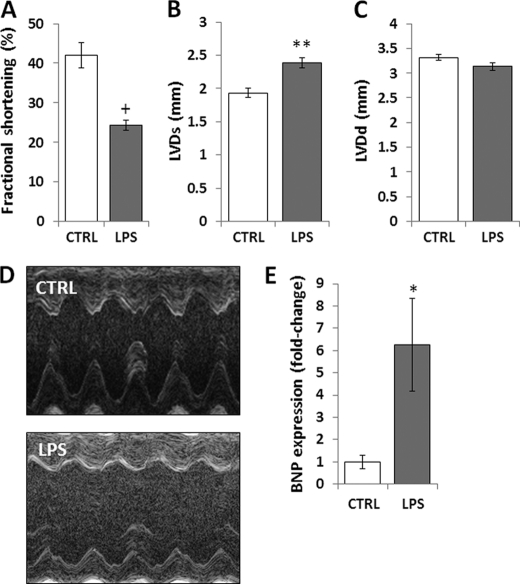
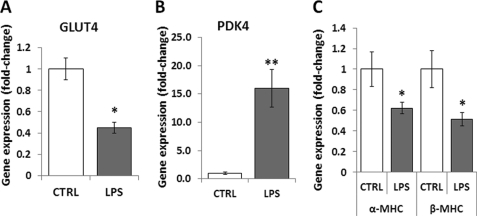
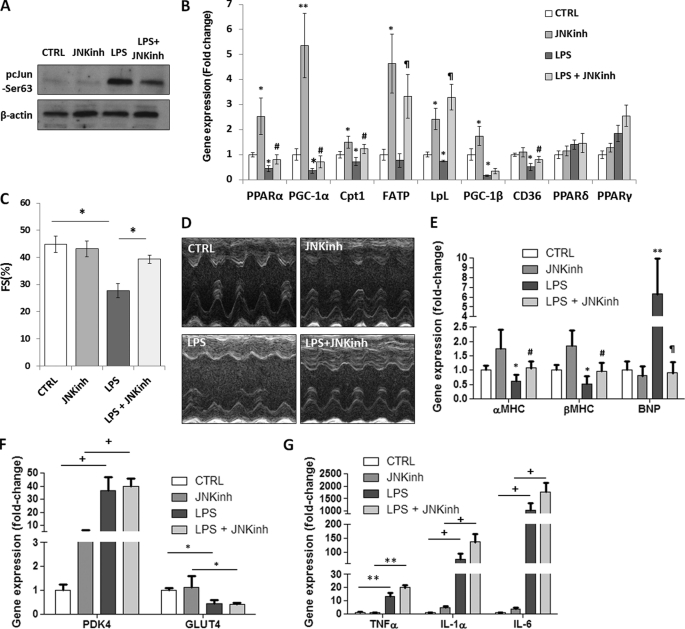
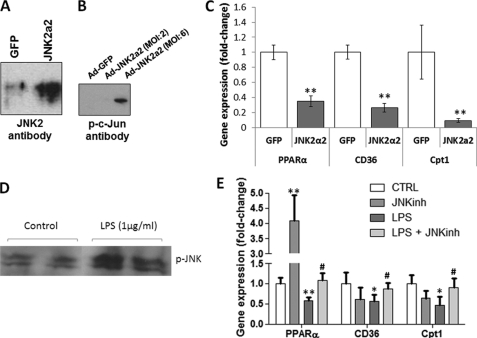
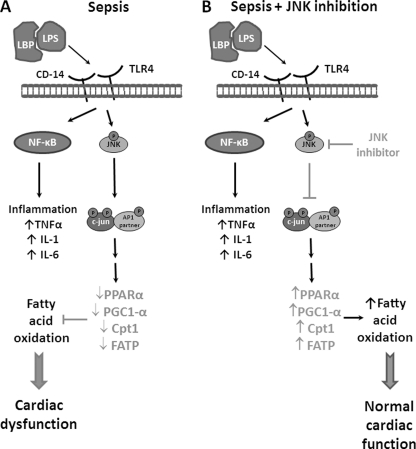
Septic shock results from bacterial infection and is associated with multi-organ failure, high mortality, and cardiac dysfunction. Sepsis causes both myocardial inflammation and energy depletion. We hypothesized that reduced cardiac energy production is a primary cause of ventricular dysfunction in sepsis. The JNK pathway is activated in sepsis and has also been implicated in impaired fatty acid oxidation in several tissues. Therefore, we tested whether JNK activation inhibits cardiac fatty acid oxidation and whether blocking JNK would restore fatty acid oxidation during LPS treatment. LPS treatment of C57BL/6 mice and adenovirus-mediated activation of the JNK pathway in cardiomyocytes inhibited peroxisome proliferator-activated receptor α expression and fatty acid oxidation. Surprisingly, none of the adaptive responses that have been described in other types of heart failure, such as increased glucose utilization, reduced αMHC:βMHC ratio or induction of certain microRNAs, occurred in LPS-treated mice. Treatment of C57BL/6 mice with a general JNK inhibitor (SP600125) increased fatty acid oxidation in mice and a cardiomyocyte-derived cell line. JNK inhibition also prevented LPS-mediated reduction in fatty acid oxidation and cardiac dysfunction. Inflammation was not alleviated in LPS-treated mice that received the JNK inhibitor. We conclude that activation of JNK signaling reduces fatty acid oxidation and prevents the peroxisome proliferator-activated receptor α down-regulation that occurs with LPS.
Figures
References
-
- Annane D., Bellissant E., Cavaillon J. M. (2005) Lancet 365, 63–78 - PubMed
-
- Levy M. M., Fink M. P., Marshall J. C., Abraham E., Angus D., Cook D., Cohen J., Opal S. M., Vincent J. L., Ramsay G. (2003) Crit. Care Med. 31, 1250–1256 - PubMed
-
- Neubauer S. (2007) N. Engl. J. Med. 356, 1140–1151 - PubMed
-
- Tessier J. P., Thurner B., Jüngling E., Lückhoff A., Fischer Y. (2003) Cardiovasc. Res. 60, 119–130 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
