

Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells
- PMID: 21873429
- PMCID: PMC3207468
- DOI: 10.1074/jbc.M111.251777
Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells
Abstract
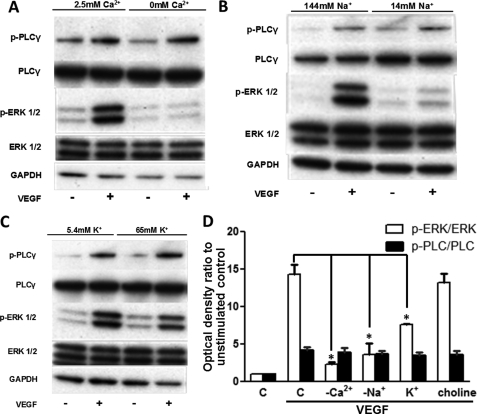
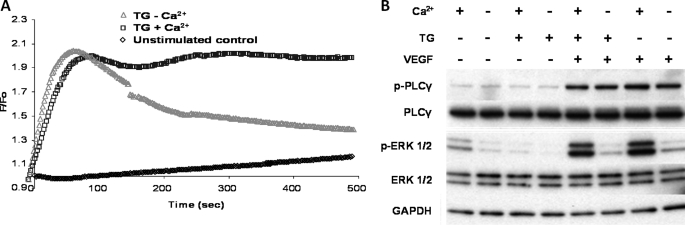
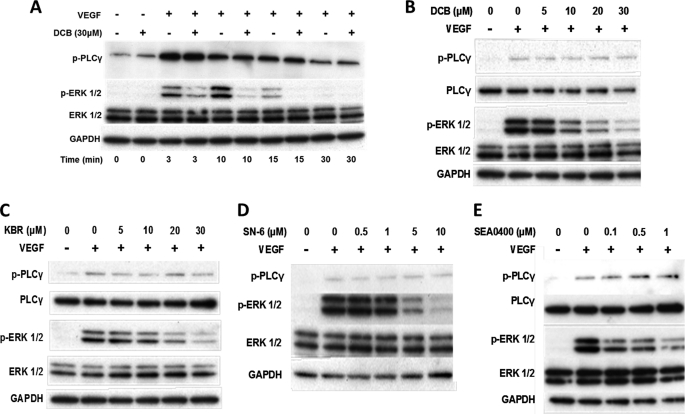
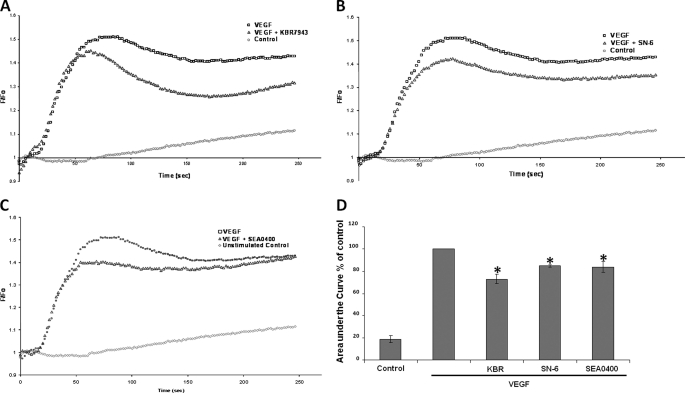
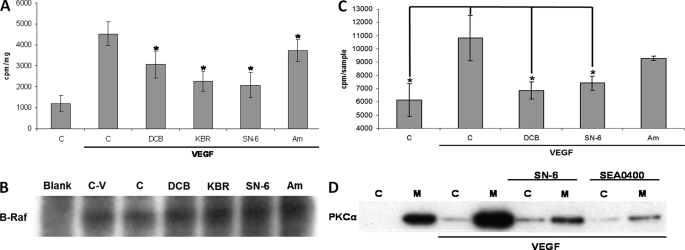
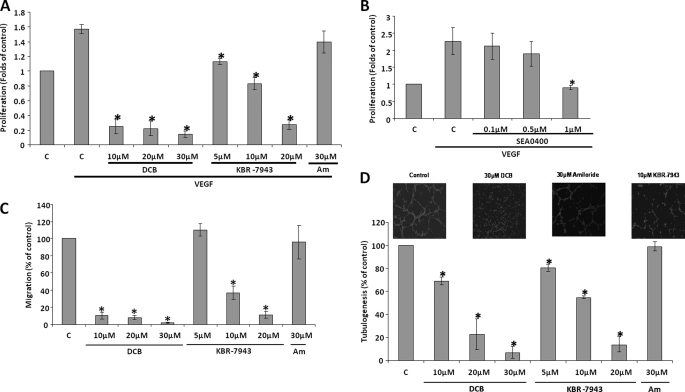
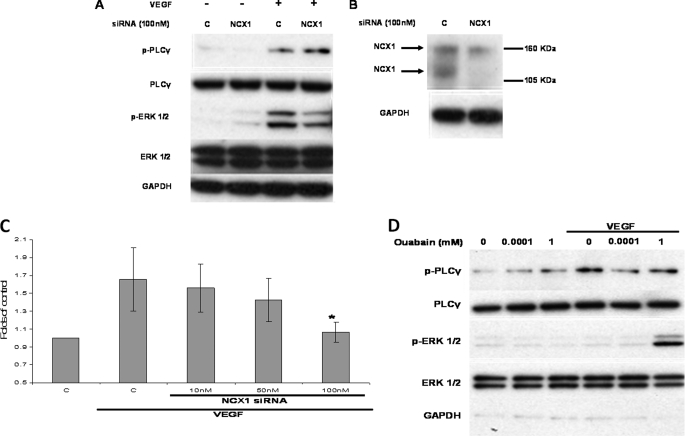
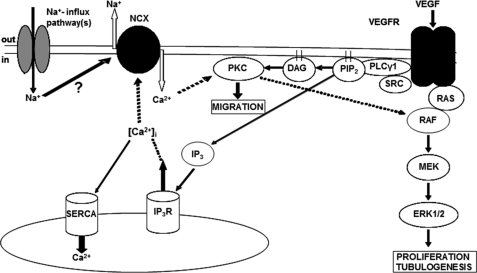
VEGF is a key angiogenic cytokine and a major target in anti-angiogenic therapeutic strategies. In endothelial cells (ECs), VEGF binds VEGF receptors and activates ERK1/2 through the phospholipase γ (PLCγ)-PKCα-B-Raf pathway. Our previous work suggested that influx of extracellular Ca(2+) is required for VEGF-induced ERK1/2 activation, and we hypothesized that this could occur through reverse mode (Ca(2+) in and Na(+) out) Na(+)-Ca(2+) exchange (NCX). However, the role of NCX activity in VEGF signaling and angiogenic functions of ECs had not previously been described. Here, using human umbilical vein ECs (HUVECs), we report that extracellular Ca(2+) is required for VEGF-induced ERK1/2 activation and that release of Ca(2+) from intracellular stores alone, in the absence of extracellular Ca(2+), is not sufficient to activate ERK1/2. Furthermore, inhibitors of reverse mode NCX suppressed the VEGF-induced activation of ERK1/2 in a time- and dose-dependent manner and attenuated VEGF-induced Ca(2+) transients. Knockdown of NCX1 (the main NCX isoform in HUVECs) by siRNA confirmed the pharmacological data. A panel of NCX inhibitors also significantly reduced VEGF-induced B-Raf activity and inhibited PKCα translocation to the plasma membrane and total PKC activity in situ. Finally, NCX inhibitors reduced VEGF-induced HUVEC proliferation, migration, and tubular differentiation in surrogate angiogenesis functional assays in vitro. We propose that Ca(2+) influx through reverse mode NCX is required for the activation and the targeting of PKCα to the plasma membrane, an essential step for VEGF-induced ERK1/2 phosphorylation and downstream EC functions in angiogenesis.
Figures
References
-
- Chung A. S., Lee J., Ferrara N. (2010) Nat. Rev. Cancer 10, 505–514 - PubMed
-
- Adams R. H., Alitalo K. (2007) Nat. Rev. Mol. Cell Biol. 8, 464–478 - PubMed
-
- Olsson A. K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006) Nat. Rev. Mol. Cell Biol. 7, 359–371 - PubMed
-
- Gerber H. P., McMurtrey A., Kowalski J., Yan M., Keyt B. A., Dixit V., Ferrara N. (1998) J. Biol. Chem. 273, 30336–30343 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
