

Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for Toll-like receptor activation and cellular signaling
- PMID: 21873432
- PMCID: PMC3196117
- DOI: 10.1074/jbc.M111.237578
Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for Toll-like receptor activation and cellular signaling
Abstract
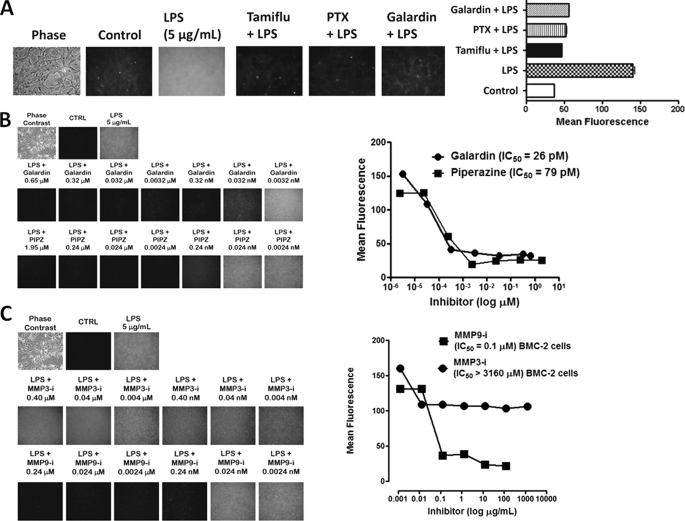
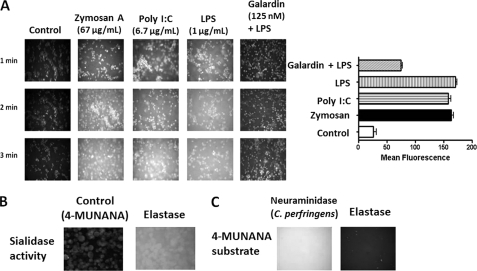
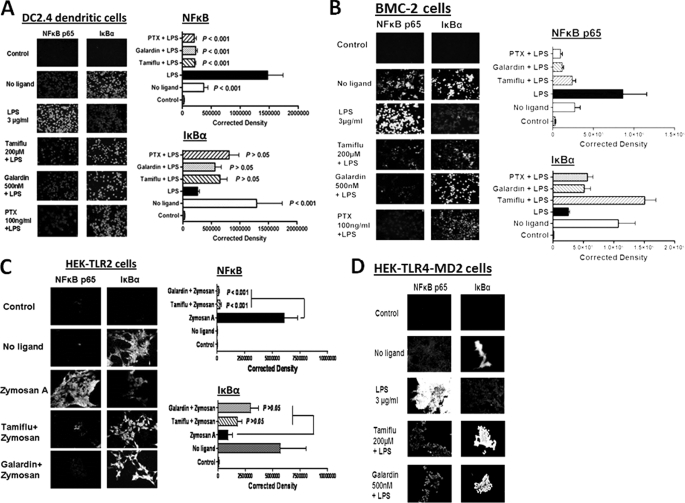
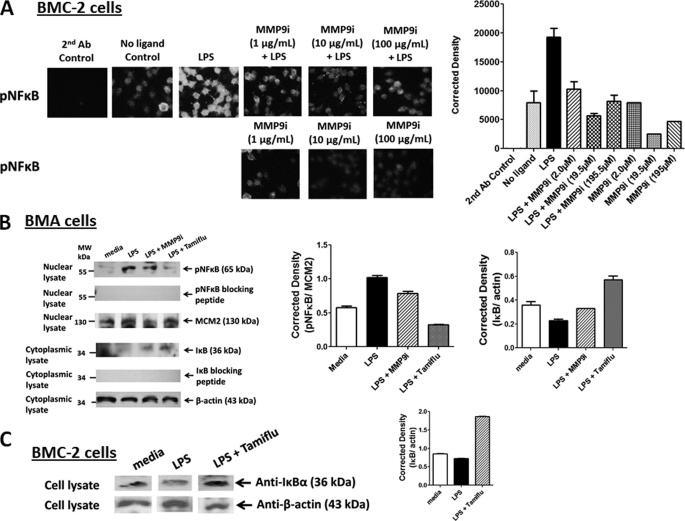
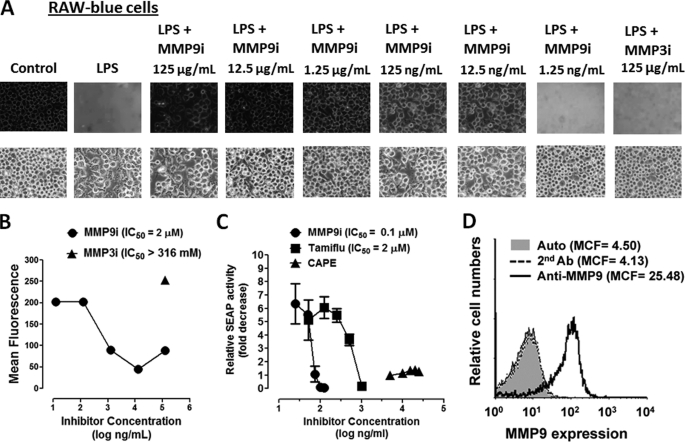
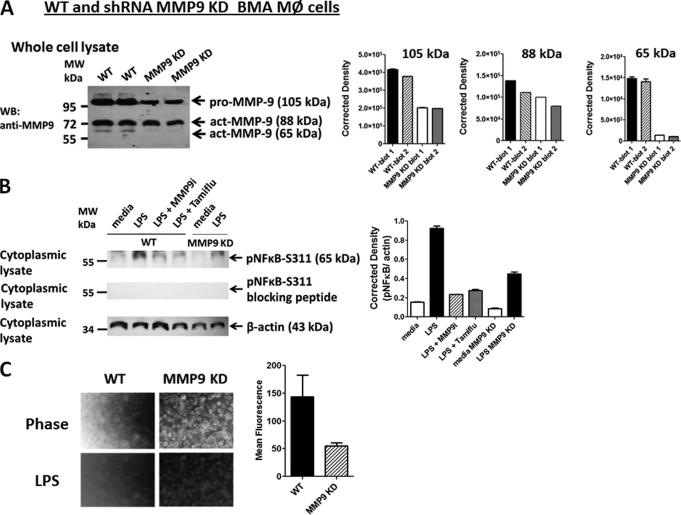
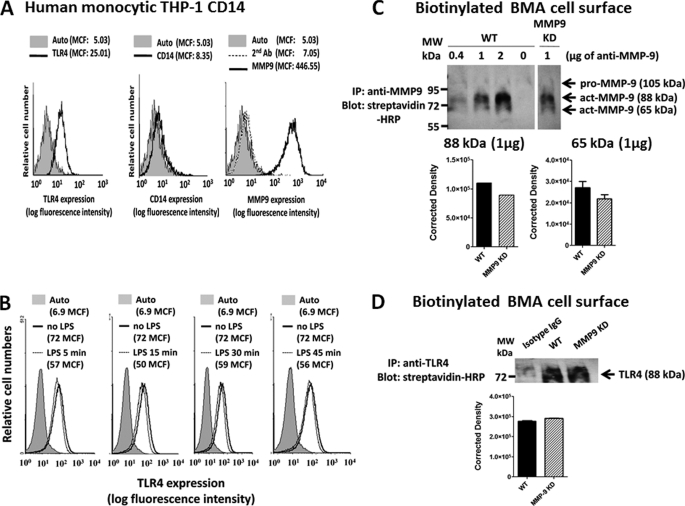
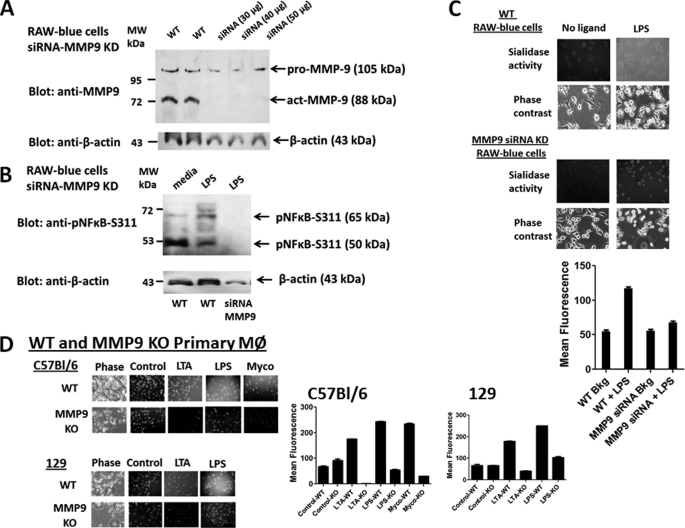
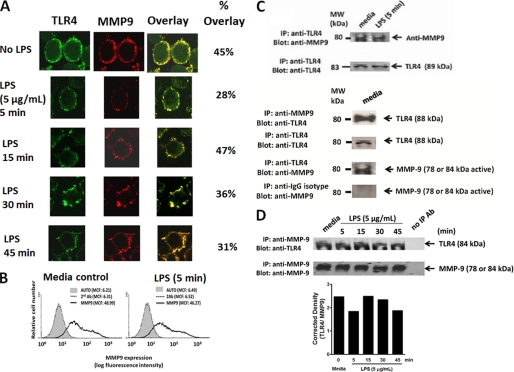
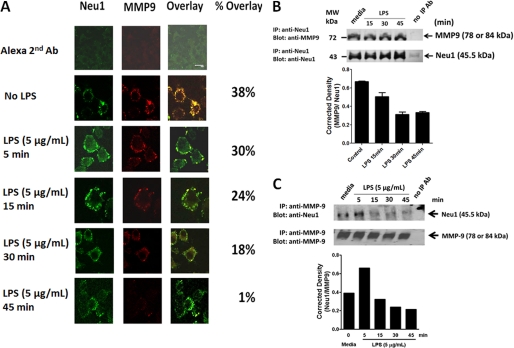
The signaling pathways of mammalian Toll-like receptors (TLRs) are well characterized, but the precise mechanism(s) by which TLRs are activated upon ligand binding remains poorly defined. Recently, we reported a novel membrane sialidase-controlling mechanism that depends on ligand binding to its TLR to induce mammalian neuraminidase-1 (Neu1) activity, to influence receptor desialylation, and subsequently to induce TLR receptor activation and the production of nitric oxide and proinflammatory cytokines in dendritic and macrophage cells. The α-2,3-sialyl residue of TLR was identified as the specific target for hydrolysis by Neu1. Here, we report a membrane signaling paradigm initiated by endotoxin lipopolysaccharide (LPS) binding to TLR4 to potentiate G protein-coupled receptor (GPCR) signaling via membrane Gα(i) subunit proteins and matrix metalloproteinase-9 (MMP9) activation to induce Neu1. Central to this process is that a Neu1-MMP9 complex is bound to TLR4 on the cell surface of naive macrophage cells. Specific inhibition of MMP9 and GPCR Gα(i)-signaling proteins blocks LPS-induced Neu1 activity and NFκB activation. Silencing MMP9 mRNA using lentivirus MMP9 shRNA transduction or siRNA transfection of macrophage cells and MMP9 knock-out primary macrophage cells significantly reduced Neu1 activity and NFκB activation associated with LPS-treated cells. These findings uncover a molecular organizational signaling platform of a novel Neu1 and MMP9 cross-talk in alliance with TLR4 on the cell surface that is essential for ligand activation of TLRs and subsequent cellular signaling.
Figures
References
-
- Amith S. R., Jayanth P., Franchuk S., Siddiqui S., Seyrantepe V., Gee K., Basta S., Beyaert R., Pshezhetsky A. V., Szewczuk M. R. (2009) Glycoconj. J. 26, 1197–1212 - PubMed
-
- Amith S. R., Jayanth P., Franchuk S., Finlay T., Seyrantepe V., Beyaert R., Pshezhetsky A. V., Szewczuk M. R. (2010) Cell. Signal. 22, 314–324 - PubMed
-
- Jayanth P., Amith S. R., Gee K., Szewczuk M. R. (2010) Cell. Signal. 22, 1193–1205 - PubMed
-
- Kovacsovics-Bankowski M., Rock K. L. (1994) Eur. J. Immunol. 24, 2421–2428 - PubMed
-
- Shen Z., Reznikoff G., Dranoff G., Rock K. L. (1997) J. Immunol. 158, 2723–2730 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
