

Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: role of oxidative stress and ceramides
- PMID: 21873444
- PMCID: PMC3233827
- DOI: 10.1152/ajplung.00385.2010
Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: role of oxidative stress and ceramides
Abstract
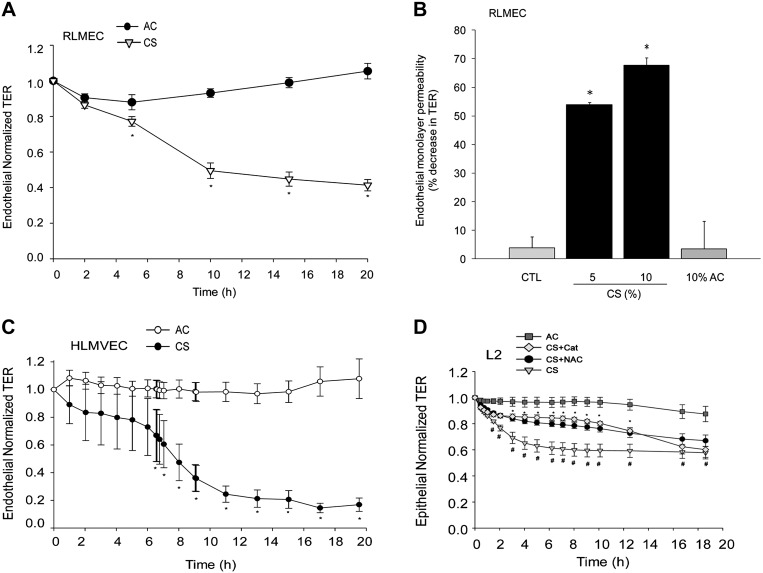
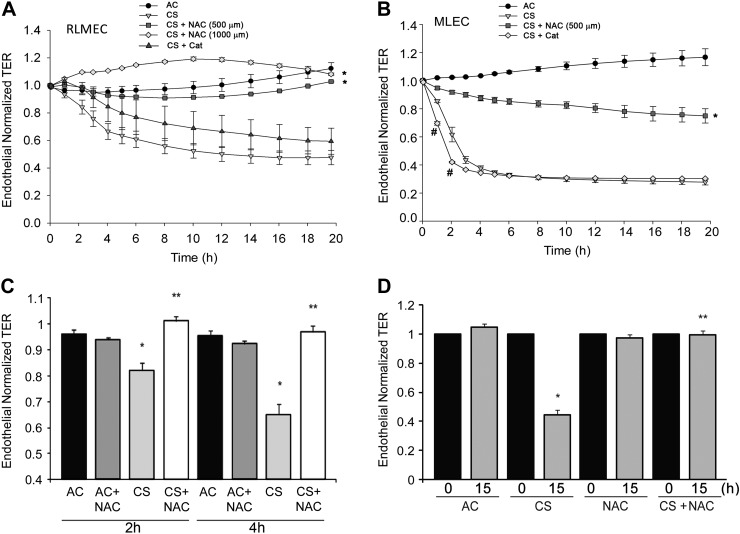
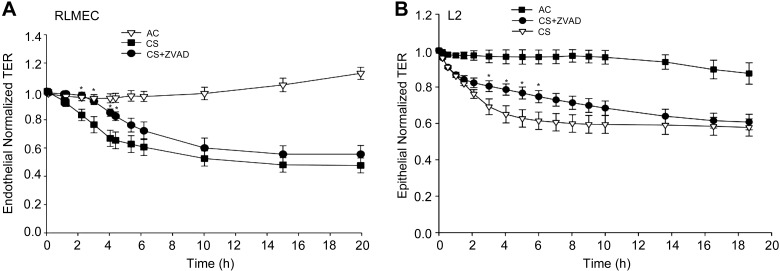
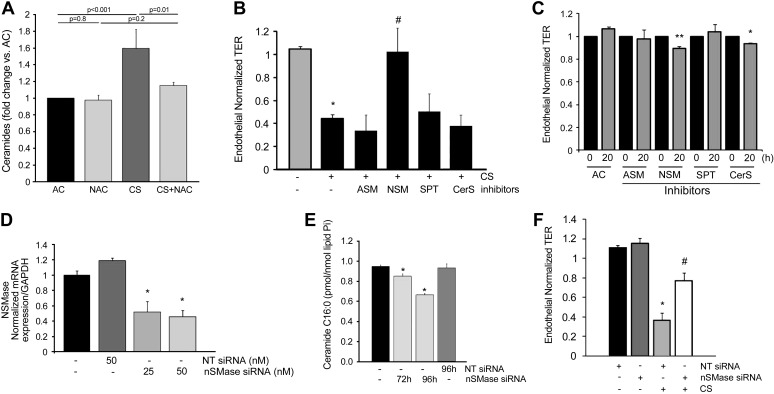
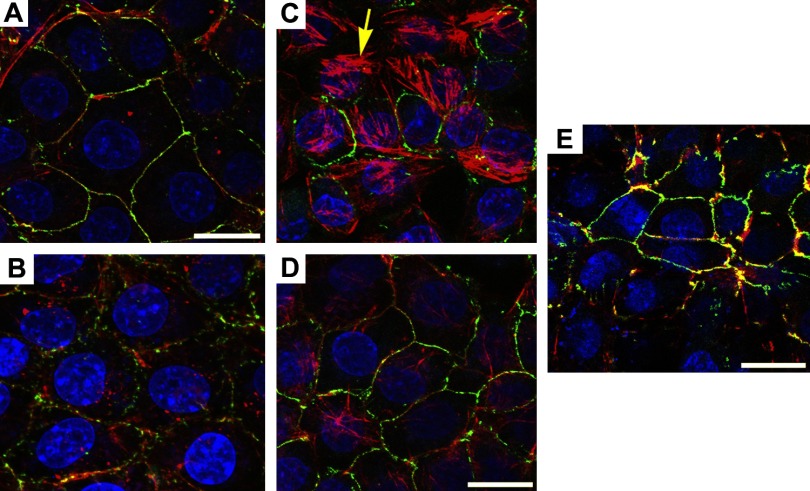
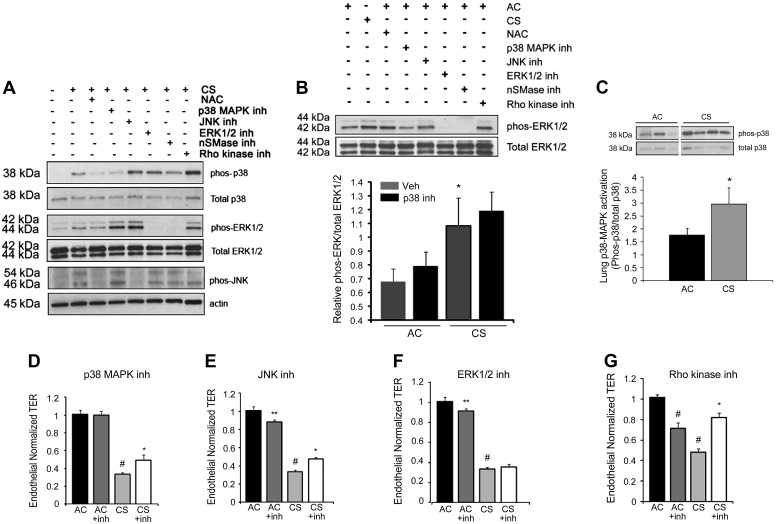
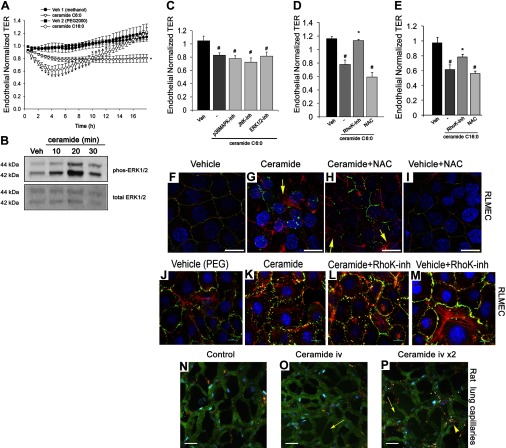
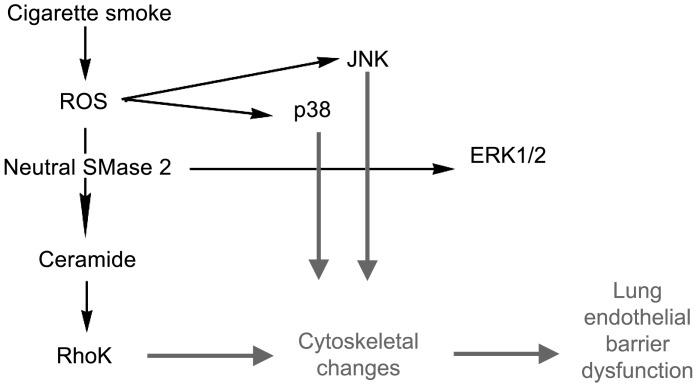
The epithelial and endothelial cells lining the alveolus form a barrier essential for the preservation of the lung respiratory function, which is, however, vulnerable to excessive oxidative, inflammatory, and apoptotic insults. Whereas profound breaches in this barrier function cause pulmonary edema, more subtle changes may contribute to inflammation. The mechanisms by which cigarette smoke (CS) exposure induce lung inflammation are not fully understood, but an early alteration in the epithelial barrier function has been documented. We sought to investigate the occurrence and mechanisms by which soluble components of mainstream CS disrupt the lung endothelial cell barrier function. Using cultured primary rat microvascular cell monolayers, we report that CS induces endothelial cell barrier disruption in a dose- and time-dependent manner of similar magnitude to that of the epithelial cell barrier. CS exposure triggered a mechanism of neutral sphingomyelinase-mediated ceramide upregulation and p38 MAPK and JNK activation that were oxidative stress dependent and that, along with Rho kinase activation, mediated the endothelial barrier dysfunction. The morphological changes in endothelial cell monolayers induced by CS included actin cytoskeletal rearrangement, junctional protein zonula occludens-1 loss, and intercellular gap formation, which were abolished by the glutathione modulator N-acetylcysteine and ameliorated by neutral sphingomyelinase inhibition. The direct application of ceramide recapitulated the effects of CS, by disrupting both endothelial and epithelial cells barrier, by a mechanism that was redox and apoptosis independent and required Rho kinase activation. Furthermore, ceramide induced dose-dependent alterations of alveolar microcirculatory barrier in vivo, measured by two-photon excitation microscopy in the intact rat. In conclusion, soluble components of CS have direct endothelial barrier-disruptive effects that could be ameliorated by glutathione modulators or by inhibitors of neutral sphingomyelinase, p38 MAPK, JNK, and Rho kinase. Amelioration of endothelial permeability may alleviate lung and systemic vascular dysfunction associated with smoking-related chronic obstructive lung diseases.
Figures
References
-
- Bartalesi B, Cavarra E, Fineschi S, Lucattelli M, Lunghi B, Martorana PA, Lungarella G. Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. Eur Respir J 25: 15–22, 2005. - PubMed
-
- Batinic-Haberle I, Cuzzocrea S, Reboucas JS, Ferrer-Sueta G, Mazzon E, Di Paola R, Radi R, Spasojevic I, Benov L, Salvemini D. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic Biol Med 46: 192–201, 2009. - PMC - PubMed
-
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Med Sci 37: 911–917, 1959. - PubMed
-
- Borbiev T, Birukova A, Liu F, Nurmukhambetova S, Gerthoffer WT, Garcia JG, Verin AD. p38 MAP kinase-dependent regulation of endothelial cell permeability. Am J Physiol Lung Cell Mol Physiol 287: L911–L918, 2004. - PubMed
-
- Boucher RC, Johnson J, Inoue S, Hulbert W, Hogg JC. The effect of cigarette smoke on the permeability of guinea pig airways. Lab Invest 43: 94–100, 1980. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous