

A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data
- PMID: 21873452
- PMCID: PMC3202289
- DOI: 10.1101/gr.119784.110
A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data
Abstract
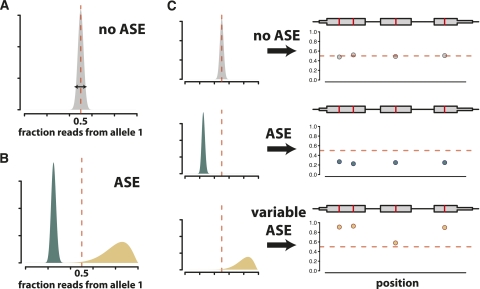
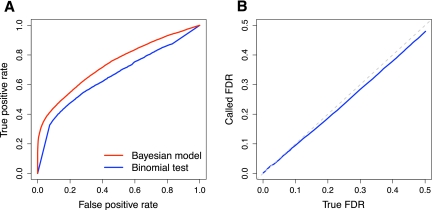
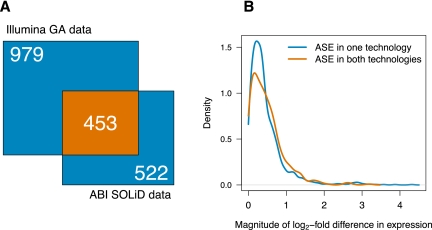
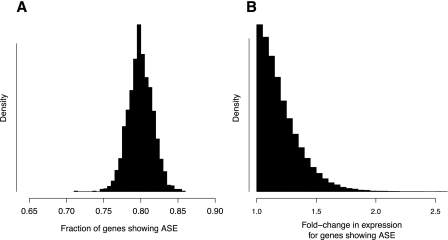
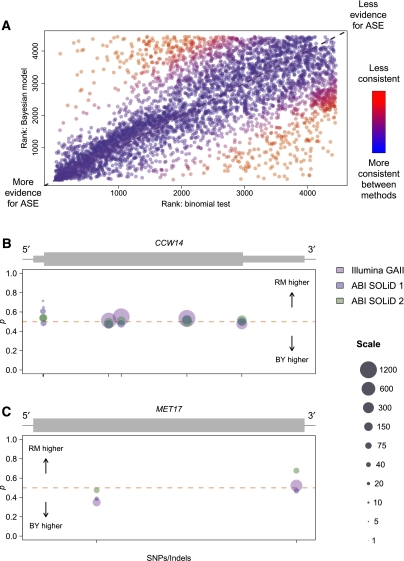
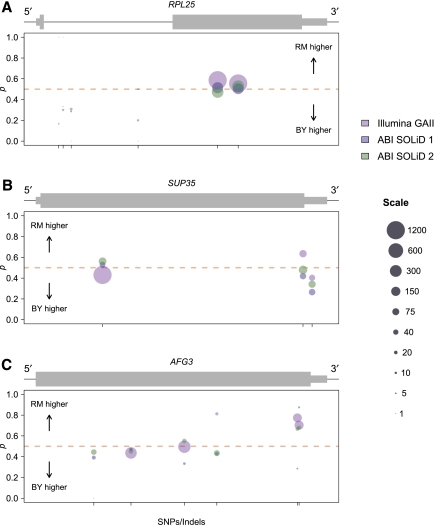
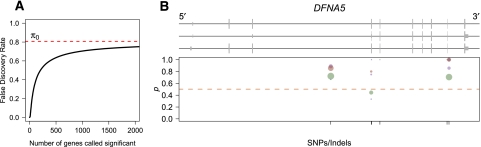
Variation in gene expression is thought to make a significant contribution to phenotypic diversity among individuals within populations. Although high-throughput cDNA sequencing offers a unique opportunity to delineate the genome-wide architecture of regulatory variation, new statistical methods need to be developed to capitalize on the wealth of information contained in RNA-seq data sets. To this end, we developed a powerful and flexible hierarchical Bayesian model that combines information across loci to allow both global and locus-specific inferences about allele-specific expression (ASE). We applied our methodology to a large RNA-seq data set obtained in a diploid hybrid of two diverse Saccharomyces cerevisiae strains, as well as to RNA-seq data from an individual human genome. Our statistical framework accurately quantifies levels of ASE with specified false-discovery rates, achieving high reproducibility between independent sequencing platforms. We pinpoint loci that show unusual and biologically interesting patterns of ASE, including allele-specific alternative splicing and transcription termination sites. Our methodology provides a rigorous, quantitative, and high-resolution tool for profiling ASE across whole genomes.
Figures
References
-
- Bray NJ, Buckland PR, Owen MJ, O'Donovan MC 2003. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum Genet 113: 149–153 - PubMed
-
- Brem RB, Yvert G, Clinton R, Kruglyak L 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 436: 701–703 - PubMed
-
- Britten RJ, Davidson EH 1971. Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. Q Rev Biol 46: 111–138 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases