

Overexpression of Nrf2 protects cerebral cortical neurons from ethanol-induced apoptotic death
- PMID: 21873460
- PMCID: PMC3228534
- DOI: 10.1124/mol.111.073262
Overexpression of Nrf2 protects cerebral cortical neurons from ethanol-induced apoptotic death
Abstract
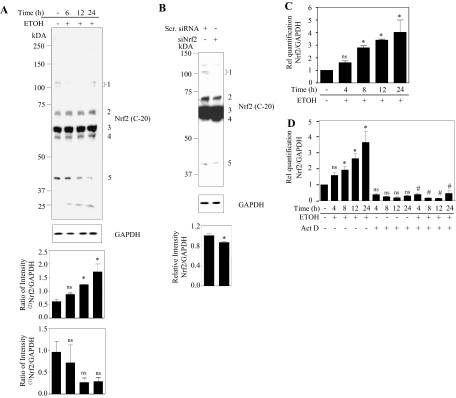
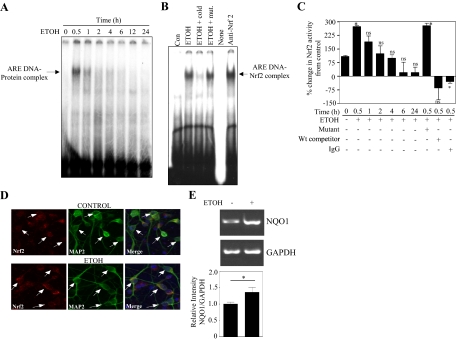
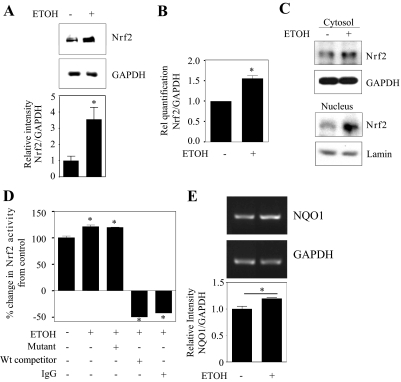
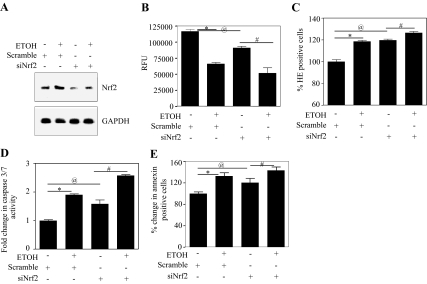
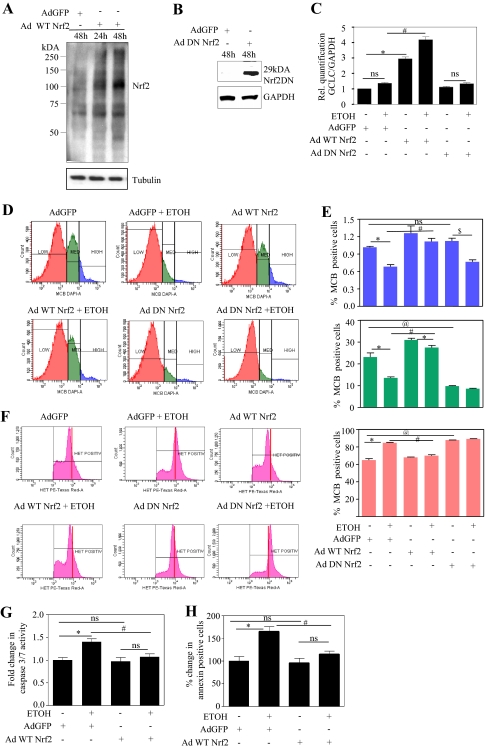
Ethanol (ETOH) can cause apoptotic death of neurons by depleting GSH with an associated increase in oxidative stress. The current study illustrates a means to overcome this ETOH-induced neurotoxicity by enhancing GSH through boosting Nrf2, a transcription factor that controls GSH homeostasis. ETOH treatment caused a significant increase in Nrf2 protein, transcript expression, Nrf2-DNA binding activity, and expression of its transcriptional target, NQO1, in primary cortical neuron (PCNs). However, this increase in Nrf2 did not maintain GSH levels in response to ETOH, and apoptotic death still occurred. To elucidate this phenomenon, we silenced Nrf2 in neurons and found that ETOH-induced GSH depletion and the increase in superoxide levels were exacerbated. Furthermore, Nrf2 knockdown resulted in significantly increased (P < 0.05) caspase 3 activity and apoptosis. Adenovirus-mediated overexpression of Nrf2 prevented ETOH-induced depletion of GSH from the medium and high GSH subpopulations and prevented ETOH-related apoptotic death. These studies illustrate the importance of Nrf2-dependent maintenance of GSH homeostasis in cerebral cortical neurons in the defense against oxidative stress and apoptotic death elicited by ETOH exposure.
Figures
Similar articles
-
Astrocytes Prevent Ethanol Induced Apoptosis of Nrf2 Depleted Neurons by Maintaining GSH Homeostasis.Open J Apoptosis. 2012 Jul;1(2):10.4236/ojapo.2012.12002. doi: 10.4236/ojapo.2012.12002. Open J Apoptosis. 2012. PMID: 24380057 Free PMC article.
-
Glutathione content as a potential mediator of the vulnerability of cultured fetal cortical neurons to ethanol-induced apoptosis.J Neurosci Res. 2008 Apr;86(5):1064-76. doi: 10.1002/jnr.21562. J Neurosci Res. 2008. PMID: 18058941 Free PMC article.
-
Programmed cell death 4 (PDCD4): a novel player in ethanol-mediated suppression of protein translation in primary cortical neurons and developing cerebral cortex.Alcohol Clin Exp Res. 2013 Jan;37(1):96-109. doi: 10.1111/j.1530-0277.2012.01850.x. Epub 2012 Jul 3. Alcohol Clin Exp Res. 2013. PMID: 22757755 Free PMC article.
-
Astrocytes protect neurons from ethanol-induced oxidative stress and apoptotic death.J Neurosci Res. 2005 Jun 1;80(5):655-66. doi: 10.1002/jnr.20502. J Neurosci Res. 2005. PMID: 15880562
-
Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress.J Neurosci. 2003 Apr 15;23(8):3394-406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. J Neurosci. 2003. PMID: 12716947 Free PMC article.
Cited by
-
Adolescent Binge Alcohol Exposure Affects the Brain Function Through Mitochondrial Impairment.Mol Neurobiol. 2018 May;55(5):4473-4491. doi: 10.1007/s12035-017-0613-4. Epub 2017 Jun 1. Mol Neurobiol. 2018. PMID: 28674997
-
Melatonin attenuates memory impairment induced by Klotho gene deficiency via interactive signaling between MT2 receptor, ERK, and Nrf2-related antioxidant potential.Int J Neuropsychopharmacol. 2014 Dec 30;18(6):pyu105. doi: 10.1093/ijnp/pyu105. Int J Neuropsychopharmacol. 2014. PMID: 25550330 Free PMC article.
-
Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells.PLoS One. 2012;7(12):e51111. doi: 10.1371/journal.pone.0051111. Epub 2012 Dec 7. PLoS One. 2012. PMID: 23236440 Free PMC article.
-
Ethanol's Effect on Coq7 Expression in the Hippocampus of Mice.Front Genet. 2018 Dec 4;9:602. doi: 10.3389/fgene.2018.00602. eCollection 2018. Front Genet. 2018. PMID: 30564271 Free PMC article.
-
Astrocytes Prevent Ethanol Induced Apoptosis of Nrf2 Depleted Neurons by Maintaining GSH Homeostasis.Open J Apoptosis. 2012 Jul;1(2):10.4236/ojapo.2012.12002. doi: 10.4236/ojapo.2012.12002. Open J Apoptosis. 2012. PMID: 24380057 Free PMC article.
References
-
- Bhave SV, Snell LD, Tabakoff B, Hoffman PL. (2000) Chronic ethanol exposure attenuates the anti-apoptotic effect of NMDA in cerebellar granule neurons. J Neurochem 75:1035–1044 - PubMed
-
- Brocardo PS, Gil-Mohapel J, Christie BR. (2011) The role of oxidative stress in fetal alcohol spectrum disorders. Brain Res Rev 67:209–225 - PubMed
-
- Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. (2002) Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 26:175–182 - PubMed
-
- Crews FT, Waage HG, Wilkie MB, Lauder JM. (1999) Ethanol pretreatment enhances NMDA excitotoxicity in biogenic amine neurons: protection by brain derived neurotrophic factor. Alcohol Clin Exp Res 23:1834–1842 - PubMed
-
- Deltour L, Ang HL, Duester G. (1996) Ethanol inhibition of retinoic acid synthesis as a potential mechanism for fetal alcohol syndrome. FASEB J 10:1050–1057 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
