

Neuronal activity modifies the DNA methylation landscape in the adult brain
- PMID: 21874013
- PMCID: PMC3183401
- DOI: 10.1038/nn.2900
Neuronal activity modifies the DNA methylation landscape in the adult brain
Abstract
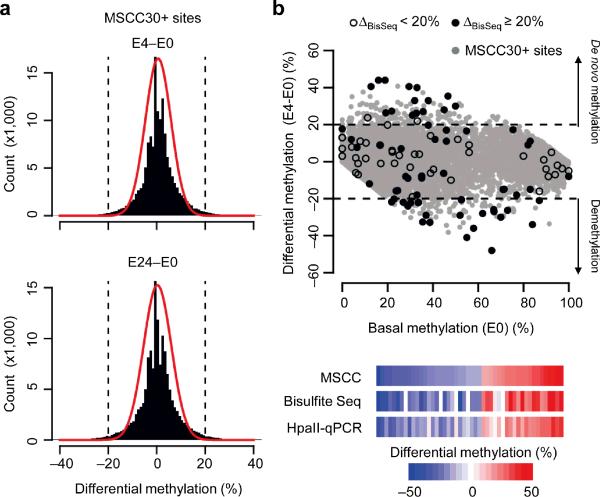
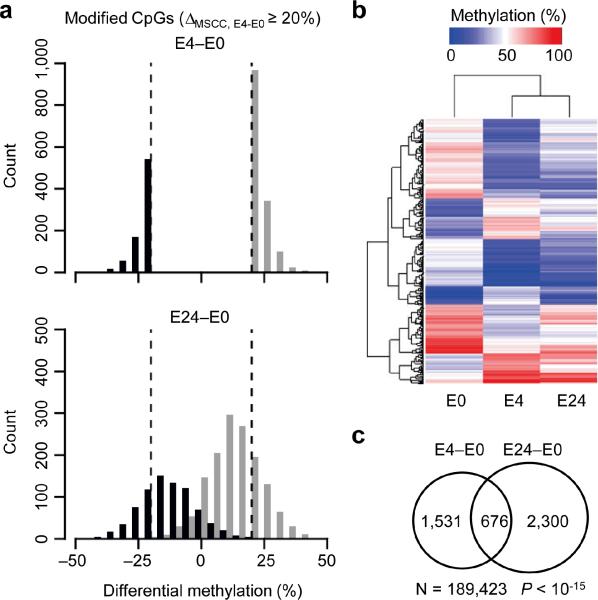
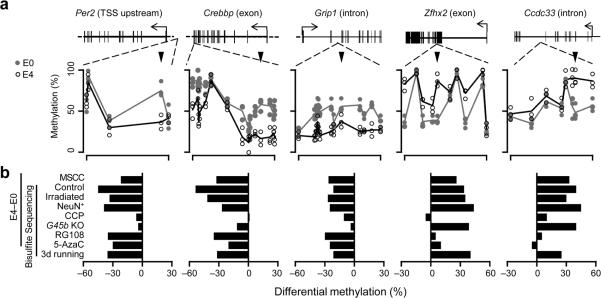
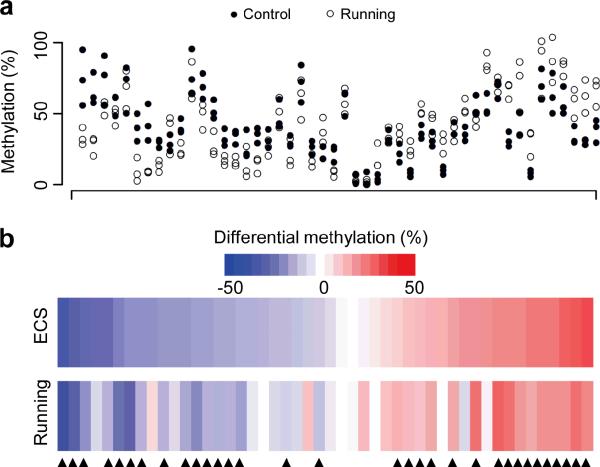
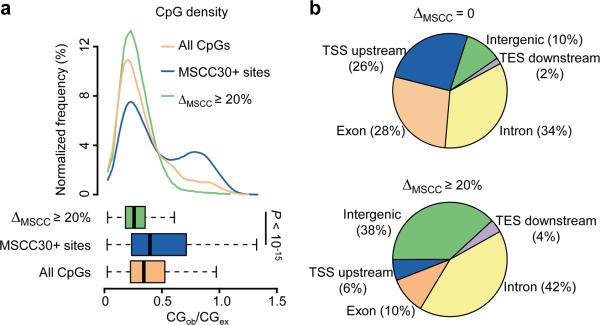
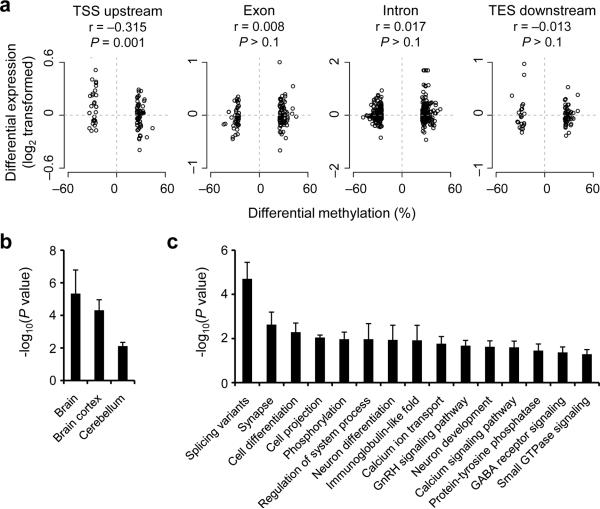
DNA methylation has been traditionally viewed as a highly stable epigenetic mark in postmitotic cells. However, postnatal brains appear to show stimulus-induced methylation changes, at least in a few identified CpG dinucleotides. How extensively the neuronal DNA methylome is regulated by neuronal activity is unknown. Using a next-generation sequencing-based method for genome-wide analysis at single-nucleotide resolution, we quantitatively compared the CpG methylation landscape of adult mouse dentate granule neurons in vivo before and after synchronous neuronal activation. About 1.4% of 219,991 CpGs measured showed rapid active demethylation or de novo methylation. Some modifications remained stable for at least 24 h. These activity-modified CpGs showed a broad genomic distribution with significant enrichment in low-CpG density regions, and were associated with brain-specific genes related to neuronal plasticity. Our study implicates modification of the neuronal DNA methylome as a previously underappreciated mechanism for activity-dependent epigenetic regulation in the adult nervous system.
© 2011 Nature America, Inc. All rights reserved.
Figures
References
-
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32. - PubMed
-
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. - PubMed
-
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–9. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
- R01 AG024984/AG/NIA NIH HHS/United States
- AG024984/AG/NIA NIH HHS/United States
- NS048271/NS/NINDS NIH HHS/United States
- R37 NS047344/NS/NINDS NIH HHS/United States
- R56 NS047344/NS/NINDS NIH HHS/United States
- R01 NS048271/NS/NINDS NIH HHS/United States
- R01 DA025779/DA/NIDA NIH HHS/United States
- K99 MH090115/MH/NIMH NIH HHS/United States
- R01 HD069184/HD/NICHD NIH HHS/United States
- R01 NS047344/NS/NINDS NIH HHS/United States
- R00 MH090115/MH/NIMH NIH HHS/United States
- HD069184/HD/NICHD NIH HHS/United States
- NS047344/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
