

N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer's disease, vascular dementia and Parkinson's disease
- PMID: 21875407
- PMCID: PMC5002349
- DOI: 10.2174/156720512801322564
N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer's disease, vascular dementia and Parkinson's disease
Abstract
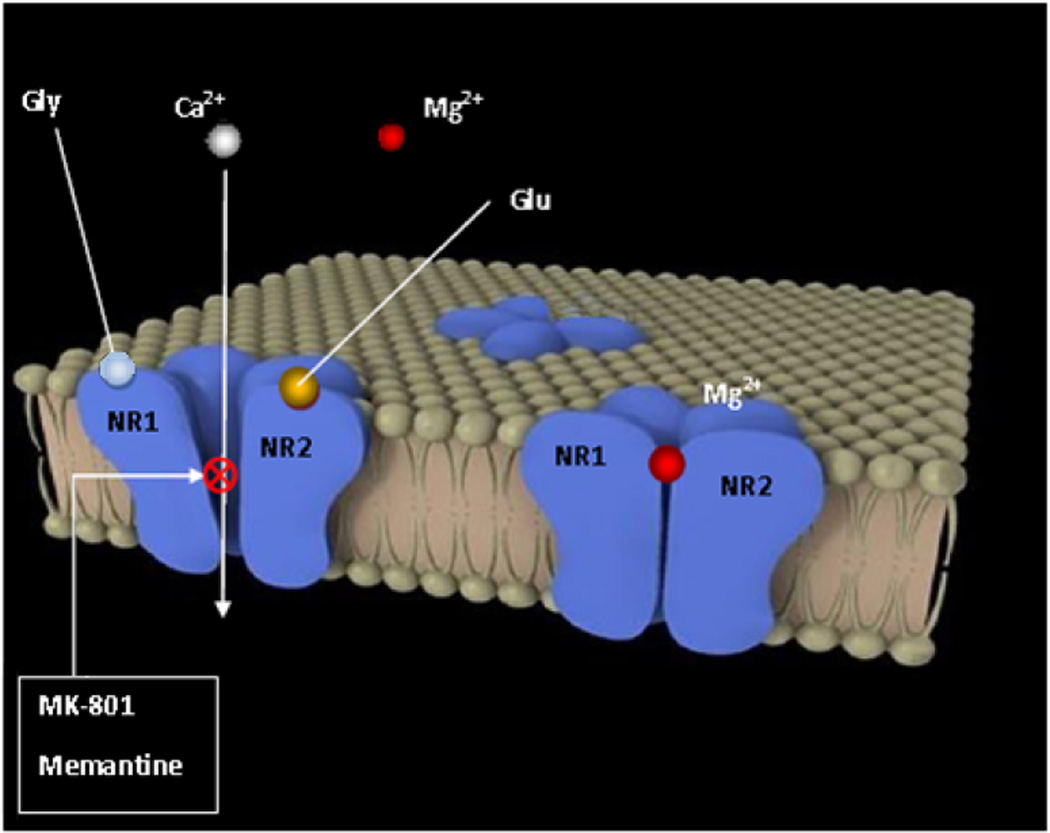
Memantine, a partial antagonist of N-methyl-D-aspartate receptor (NMDAR), approved for moderate to severe Alzheimer's disease (AD) treatment within the U.S. and Europe under brand name Namenda (Forest), Axura and Akatinol (Merz), and Ebixa and Abixa (Lundbeck), may have potential in alleviating additional neurological conditions, such as vascular dementia (VD) and Parkinson's disease (PD). In various animal models, memantine has been reported to be a neuroprotective agent that positively impacts both neurodegenerative and vascular processes. While excessive levels of glutamate result in neurotoxicity, in part through the over-activation of NMDARs, memantine-as a partial NMDAR antagonist, blocks the NMDA glutamate receptors to normalize the glutamatergic system and ameliorate cognitive and memory deficits. The key to memantine's therapeutic action lies in its uncompetitive binding to the NMDAR through which low affinity and rapid off-rate kinetics of memantine at the level of the NMDAR-channel preserves the physiological function of the receptor, underpinning memantine's tolerability and low adverse event profile. As the biochemical pathways evoked by NMDAR antagonism also play a role in PD and since no other drug is sufficiently effective to substitute for the first-line treatment of L-dopa despite its side effects, memantine may be useful in PD treatment with possibly fewer side effects. In spite of the relative modest nature of its adverse effects, memantine has been shown to provide only a moderate decrease in clinical deterioration in AD and VD, and hence efforts are being undertaken in the design of new and more potent memantine-based drugs to hopefully provide greater efficacy.
Conflict of interest statement
The authors declare that they do not have any conflicts of interest.
Figures

References
-
- Care TSCoTAiH. Dementia - Etiology and Epidemiology. A Systematic Review. 2008;1:755.
-
- Lahiri DK, Rogers JT, Greig NH, Sambamurti K. Rationale for the development of cholinesterase inhibitors as anti-Alzheimer agents. Curr Pharm Des. 2004;10(25):3111–3119. - PubMed
-
- Delacourte A. [From physiopathology to treatment of Alzheimer’s disease] Rev Neurol (Paris) 2006;162(10):909–912. - PubMed
-
- Joray S, Herrmann F, Mulligan R, Schnider A. Mechanism of disorientation in Alzheimer’s disease. Eur Neurol. 2004;52(4):193–197. - PubMed
-
- Esposito F, Rochat L, Van der Linden AC, Lekeu F, Quittre A, Charnallet A, et al. Apathy and executive dysfunction in Alzheimer disease. Alzheimer Dis Assoc Disord. 2010;24(2):131–137. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
