

WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity
- PMID: 21875954
- PMCID: PMC3182055
- DOI: 10.1084/jem.20110200
WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity
Abstract
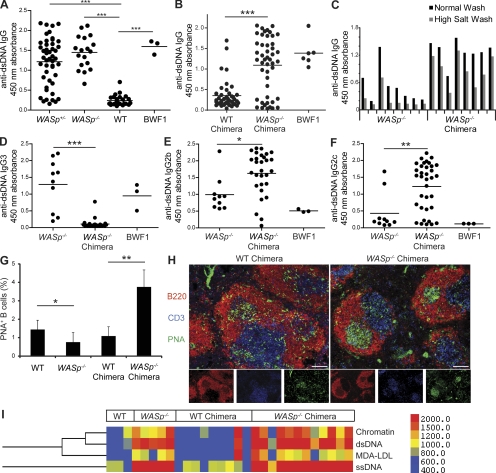
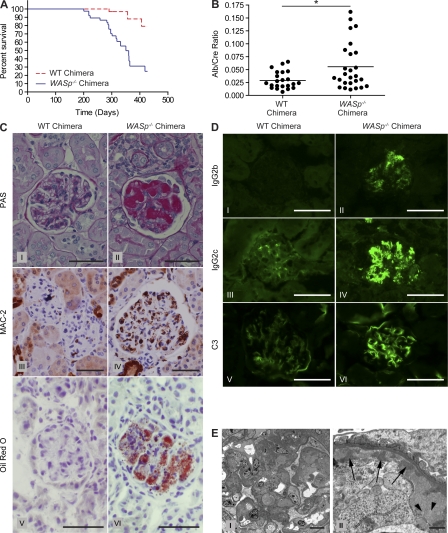
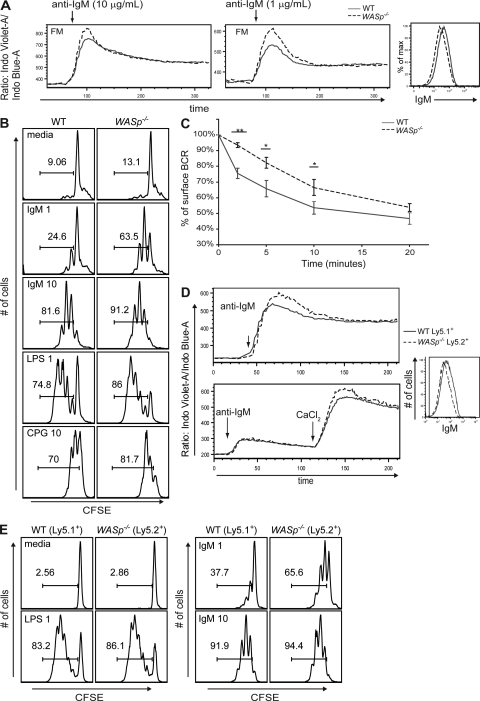
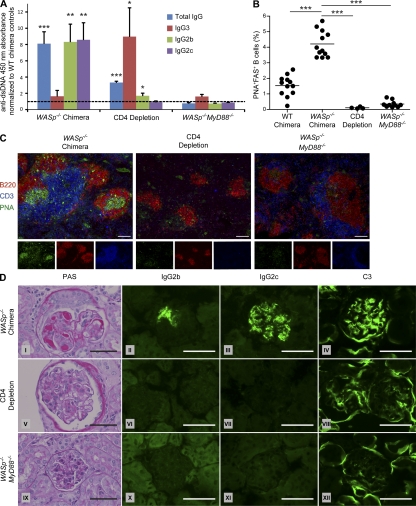
Patients with the immunodeficiency Wiskott-Aldrich syndrome (WAS) frequently develop systemic autoimmunity. Here, we demonstrate that mutation of the WAS gene results in B cells that are hyperresponsive to B cell receptor and Toll-like receptor (TLR) signals in vitro, thereby promoting a B cell-intrinsic break in tolerance. Whereas this defect leads to autoantibody production in WAS protein-deficient (WASp(-/-)) mice without overt disease, chimeric mice in which only the B cell lineage lacks WASp exhibit severe autoimmunity characterized by spontaneous germinal center formation, class-switched autoantibodies, renal histopathology, and early mortality. Both T cell help and B cell-intrinsic TLR engagement play important roles in promoting disease in this model, as depletion with anti-CD4 antibodies or generation of chimeric mice with B cells deficient in both WASp and MyD88 prevented development of autoimmune disease. These data highlight the potentially harmful role for cell-intrinsic loss of B cell tolerance in the setting of normal T cell function, and may explain why WAS patients with mixed chimerism after stem cell transplantation often develop severe humoral autoimmunity.
Figures
Comment in
-
B cells: WASP stings autoreactive B cells.Nat Rev Immunol. 2011 Sep 16;11(10):642. doi: 10.1038/nri3074. Nat Rev Immunol. 2011. PMID: 21921922 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
