

Review
doi: 10.1101/cshperspect.a007526.
Protein folding and quality control in the ER
Affiliations
- PMID: 21875985
- PMCID: PMC3220362
- DOI: 10.1101/cshperspect.a007526
Item in Clipboard
Review
Protein folding and quality control in the ER
Cold Spring Harb Perspect Biol.
.
Erratum in
-
Protein folding and quality control in the ER.Cold Spring Harb Perspect Biol. 2012 Aug 1;4(8):a015438. doi: 10.1101/cshperspect.a015438. Cold Spring Harb Perspect Biol. 2012. PMID: 22855729 Free PMC article. Review. No abstract available.
Abstract
The endoplasmic reticulum (ER) uses an elaborate surveillance system called the ER quality control (ERQC) system. The ERQC facilitates folding and modification of secretory and membrane proteins and eliminates terminally misfolded polypeptides through ER-associated degradation (ERAD) or autophagic degradation. This mechanism of ER protein surveillance is closely linked to redox and calcium homeostasis in the ER, whose balance is presumed to be regulated by a specific cellular compartment. The potential to modulate proteostasis and metabolism with chemical compounds or targeted siRNAs may offer an ideal option for the treatment of disease.
Figures
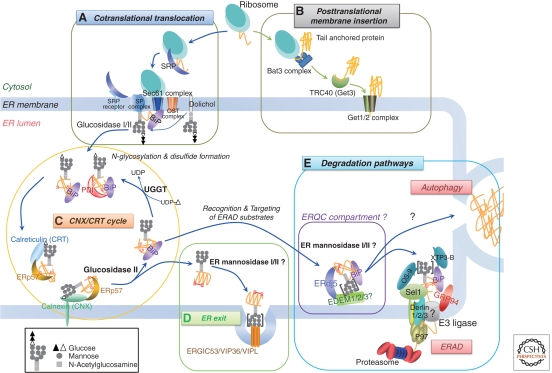
Schematic model of the ER quality control system. Selected components are depicted in this model. (A) Cotranslational translation: The ER signal sequence of a newly synthesized polypeptide is bound by a signal-recognition particle (SRP). The SRP–ribosome complex is guided to the Sec complex by an ER membrane-localized SRP receptor. After release of the SRP and SRP receptor, the polypeptide begins translocation. Subsequently, the signal sequence is processed by the signal peptidase (SP) complex (Paetzel et al. 2002). Transfer of oligosaccharides is catalyzed by the oligosaccharyl transferase (OST) complex, and the two outermost glucose residues are sequentially removed by glucosidases I and II. (B) Posttranslational membrane insertion: a tail-anchored (TA) protein is posttranslationally inserted into the ER membrane. The carboxy-terminal single trans-membrane domain of the TA protein is recognized by the Bat3 complex and transferred to the cytoplasmic chaperone TRC40 for targeting to the ER-membrane localized Get1/2 receptor. (C) CNX/CRT cycle: a monoglucosylated N-glycan of the polypeptide recruits the ER lectin chaperones, calnexin (CNX) and/or calreticulin (CRT), which promote proper folding by preventing aggregation and premature export from the ER. ERp57, a CNX/CRT-bound oxidoreductase, catalyzes disulfide formation. Trimming of the innermost glucose residue by glucosidase II then releases the polypeptide from CNX/CRT. UDP-glucose/glycoprotein glucosyl transferase (UGGT) monitors the folding state of released glycoprotein and, if the correct conformation has not been achieved, UGGT reglucosylates it to be reengaged by CNX/CRT. (D) ER exit: The natively folded protein is released from the CNX/CRT cycle and transported to its destination. In the early secretory pathway, lectins (ERGIC53, VIP36, and VIPL) support the sorting or trafficking of glycosylated proteins from the ER to the Golgi (Kamiya et al. 2008; Dancourt and Barlowe 2010). “[ ]” indicates that there are several possibilities for N-glycan formation. (E) Degradation pathways: Terminally misfolded proteins are degraded primarily through ER-associated degradation (ERAD) or autophagic degradation. Before degradation, N-glycans on ERAD substrates are extensively trimmed for efficient degradation, possibly in a specific compartment within the ER known as the ER quality control (ERQC) compartment, where ERAD machineries such as ER mannosidase I and EDEM family proteins are enriched. Subsequently, ERAD substrates are retrotranslocated into the cytosol, possibly through an E3 ligase complex, and finally degraded by the ubiquitin proteasome pathway. A recent report suggests that misfolded TA proteins are also degraded by the ERAD pathway (Claessen et al. 2010). Autophagy also degrades some ERAD substrates, but its recognition mechanism is not well understood.

Schematic models of mammalian E3 ligase complexes. Selected components are depicted in each model (Tsai and Weissman 2010). The majority of motif annotations are taken from the Pfam and SMART databases (see Tables 1 and 2; Letunic et al. 2009; Finn et al. 2010). (A) HRD1 ligase complex: This complex is a well-illustrated E3 complex that primarily targets proteins for ERAD-L. OS-9 and XTP3-B recognize aberrant nonglycosylated or glycosylated proteins in the ER lumen. Both associate with the HRD1 complex through SEL1L in a mutually exclusive manner. BiP and GRP94 presumably cooperate to regulate the assembly/disassembly of the HRD1 complex and sequester misfolded proteins to prevent other interactions until retrotranslocation. Derlin family proteins (Derlin 1, 2, or 3) might participate in substrate retrotranslocation from the ER lumen into the cytosol. UBXD8 and UBXD2 bind to p97/VCP through their UBX domain and accelerate the degradation of ERAD substrates. E2 ubiquitin-conjugating enzymes (Ube2j1, Ube2k) mediate substrate ubiquitination (Burr et al. 2011), whereas the p97/VCP hexamer promotes substrate extraction into the cytosol. Ubiquilin-1 is suggested to act as a ubiquitin–proteasome shuttle protein. Other ubiquitin-chain modifiers may also come into play, such as E4 ubiquitin-conjugating enzyme (Ufd2), Ufd2 inhibitor (Ufd3), and deubiquitinases (Ataxin-3, YOD1, VCIP135) (Rumpf and Jentsch 2006). The deglycosylating enzyme PNGase releases N-linked glycan chains from the glycopeptide (Tanabe et al. 2006). The proteasome then captures the polyubiquitin chains on the substrate through specific subunits (Rpn10/13, Rpt5) and degrades it. (B) RMA1 ligase complex: RMA1/RNF5 associates with Derlin-1 and E2 Ube2j1. BAP31, known to be an ER sorting factor of diverse membrane proteins, interacts with RMA1 as well as components of the Sec61 pore, which suggests that BAP31 might recruit the ERAD complex to the translocon channel to clear newly synthesized misfolded membrane proteins from the channel. In addition, DNAJB12, which contains the cytosolic J-domain, may also participate in the degradation of membrane proteins together with HSP70. All of these factors have been reported to play a role in the degradation of cystic fibrosis trans-membrane conductance regulator (CFTR) and its mutant (CFTRΔ508) during the early steps of translation (Younger et al. 2006; Morito et al. 2008). (C) Cytosolic E3 ligases: CHIP (carboxyl terminus of Hsp70-interacting protein) possesses a U-box domain, which has a structure similar to the RING-finger domain, and a tetratricopeptide repeat domain (TPR) that interacts with Hsp70 and Hsp90. In contrast to RMA1, CHIP posttranslationally monitors the folding of CFTR or CFTRΔ508. Parkin, which is responsible for autosomal recessive juvenile Parkinsonism, targets several proteins, such as O-glycosylated α-synuclein, the Pael receptor (Pael-R), Synphilin-1, and Tau. Regarding Pael-R, Parkin and CHIP act together to enhance its ubiquitination and inhibit cell death induced by accumulated unfolded Pael-R. Parkin also works with the E2 proteins Ube2j1 and Ube2g2, which suggests that it is involved in ERAD. The SCF (SKP1-CUL1-F-box protein) is composed of three proteins (Cullin1, Skp1, and RING finger protein Rbx1) and one F-box protein (Fbs1 or Fbs2). Fbs1 and Fbs2 are novel F-box proteins that recognize sugar chains in N-linked glycoproteins and show a chaperone-like activity to prevent their aggregation (Yoshida and Tanaka 2010). (D) gp78 ligase complex: The carboxyl terminus of gp78 is composed of four motifs: RING, CUE, G2BR (Ube2g2 binding region), and VIM (p97/VCP-interacting motif). The gp78 ligase complex usually consists of Ube2g2, Derlin-1, p97/VCP, and UBXD2 or UBXD8. gp78 mediates several ERAD substrates, including T-cell receptor subunits (CD3-δ and TCR-α), apolipoprotein B-100, Insig-1, and HMG-CoA reductase. The latter two substrates suggest that gp78 is involved in cholesterol metabolism (Jo and Debose-Boyd 2010). gp78 also cooperates with RMA1 to degrade CFTRΔ508. SVIP is reported to inhibit the assembly of the gp78 ligase complex (Derlin-1, gp78, and p97), which suggests an inhibitory effect on ERAD (Ballar et al. 2007). (E) TEB4 (Doa10) ligase complex: TEB4 is known to be a mammalian homolog of yeast Doa10. Together with Ube2g2, TEB4 is implicated in the degradation of ER resident type 2 iodothyronine deiodinase (Zavacki et al. 2009). Based on the homology of the yeast Doa10, TEB4 might also interact with Ube2j1 (Ubc6) and UBXD8 (Ubx2).

ER redox and calcium homeostasis on the MAM. On the mitochondria-associated membrane (MAM), ER chaperones (CNX, BiP), oxidoreductases (Ero1α, ERp44), and Ca2+ channels/pumps (IP3R3, SERCAs, and Sig-1R) are enriched, thereby creating an ideal environment for oxidative protein folding, as well as the regulation of Ca2+ flux. (A) ER redox homeostasis: Ero1α serves as the primary oxidase of PDI. Concomitantly, hydrogen peroxide (H2O2) is thought to be produced from oxygen as an electron acceptor. Peroxiredoxin IV (PRDX4) is thought to work as a H2O2 reducer. PRDX4 can also oxidize some PDI family members. Oxidized PDI family members drive oxidative protein folding, as well as oxidize GSH into GSSG, thereby generating an oxidative environment. The FAD, cofactor of Ero1α, may be delivered from mitochondria via unknown transporters. (B) Calcium flux: Mammalian cells contain two main channels responsible for Ca2+ efflux from the ER, inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs), and one pump responsible for Ca2+ influx into the ER, sarcoplasmic reticulum Ca2+-ATPase (SERCAs). ERp44 interacts with the luminal loop of IP3R type 1 (IP3R1) and inhibits its activity. ERp57 oxidizes the luminal thiols of SERCA2b in a Ca2+-dependent manner (Li and Camacho 2004). By-product of oxidative protein folding (i.e., ROS) affects the redox states of the channels (RyR and IP3R1) and changes their activities. In this way redox homeostasis and Ca2+ flux are interrelated. Sigma-1 receptor (Sig-1R) works as a Ca2+ sensor and interacts with BiP. On Ca2+ depletion from the ER via IP3R, BiP dissociates from Sig-1R, interacts with IP3R, and protects intrinsically unstable IP3R from degradation. Cytosolic sorting protein PACS-2 recruits CNX to the MAM (Myhill et al. 2008). Both CRT and CNX interact with SERCA2b and inhibit Ca2+ oscillations (John et al. 1998). The MAM is also the place where energy (ATP) and lipids are exchanged between ER and mitochondria. Ca2+ and ATP levels affect the activities of ER chaperones and foldases. Thus, redox homeostasis, Ca2+ flux and the activities of ER foldases integrally affect the oxidative protein folding capacity in the ER.
References
-
- Aebi M, Bernasconi R, Clerc S, Molinari M 2010. N-glycan structures: Recognition and processing in the ER. Trends Biochem Sci 35: 74–82 - PubMed
-
- Appenzeller-Herzog C 2011. Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. J Cell Sci 124: 847–855 - PubMed
-
- Appenzeller-Herzog C, Ellgaard L 2008. The human PDI family: Versatility packed into a single fold. Biochim Biophys Acta 1783: 535–548 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources