

Helicobacter pylori arginase mutant colonizes arginase II knockout mice
- PMID: 21876618
- PMCID: PMC3160534
- DOI: 10.3748/wjg.v17.i28.3300
Helicobacter pylori arginase mutant colonizes arginase II knockout mice
Abstract
Aim: To investigate the role of host and bacterial arginases in the colonization of mice by Helicobacter pylori (H. pylori).
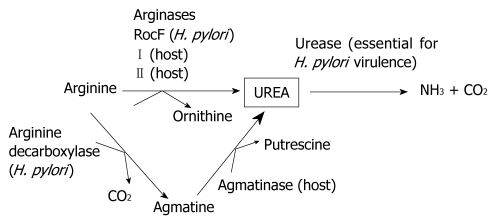
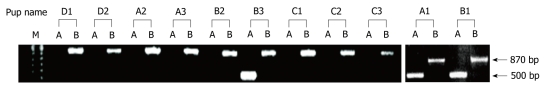
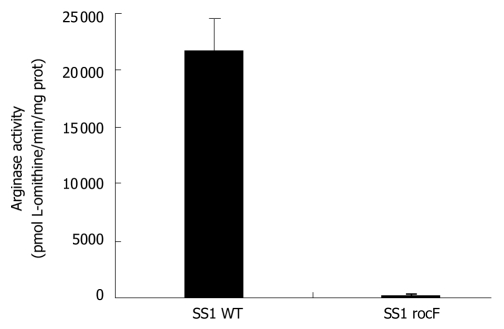
Methods: H. pylori produces a very powerful urease that hydrolyzes urea to carbon dioxide and ammonium, which neutralizes acid. Urease is absolutely essential to H. pylori pathogenesis; therefore, the urea substrate must be in ample supply for urease to work efficiently. The urea substrate is most likely provided by arginase activity, which hydrolyzes L-arginine to L-ornithine and urea. Previous work has demonstrated that H. pylori arginase is surprisingly not required for colonization of wild-type mice. Hence, another in vivo source of the critical urea substrate must exist. We hypothesized that the urea source was provided by host arginase II, since this enzyme is expressed in the stomach, and H. pylori has previously been shown to induce the expression of murine gastric arginase II. To test this hypothesis, wild-type and arginase (rocF) mutant H. pylori strain SS1 were inoculated into arginase II knockout mice.
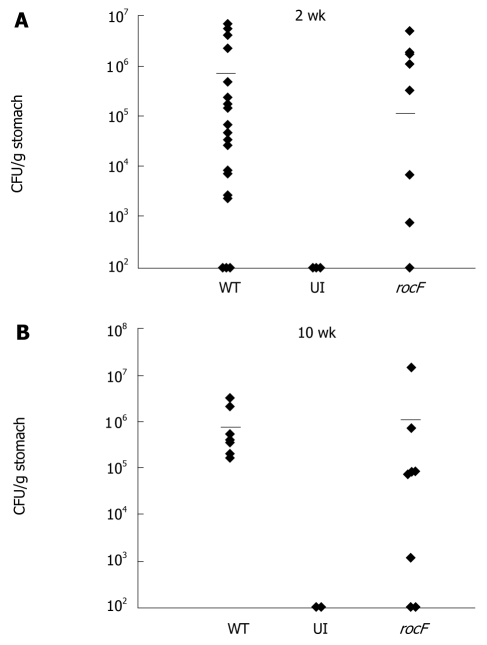
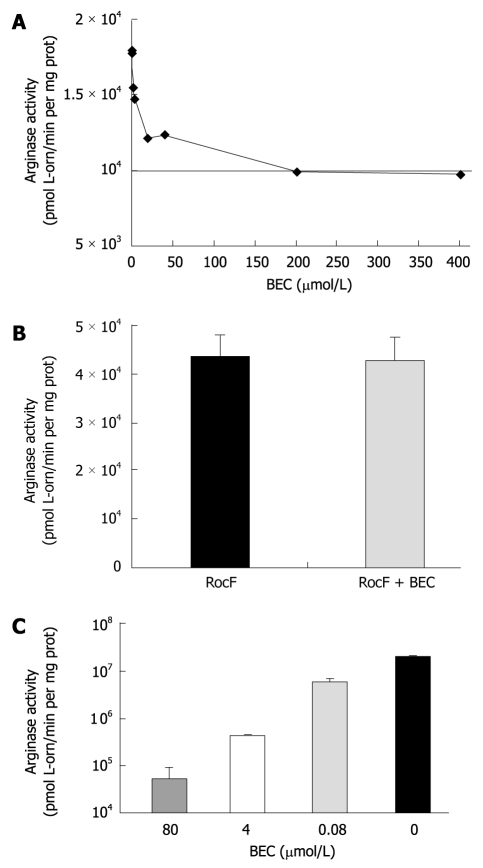
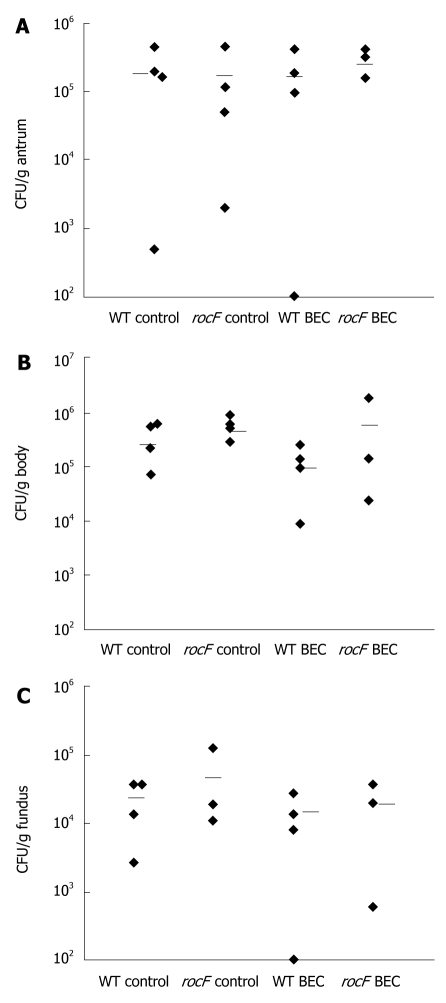
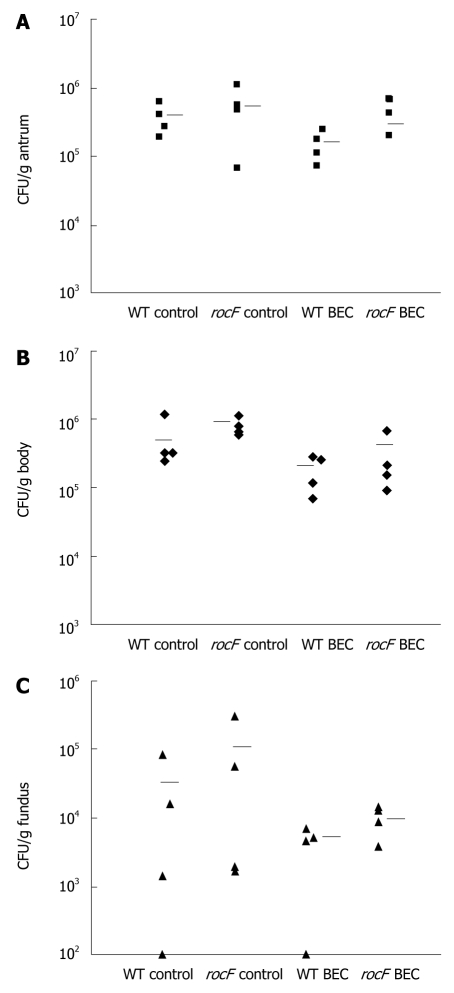
Results: Surprisingly, both the wild-type and rocF mutant bacteria still colonized arginase II knockout mice. Moreover, feeding arginase II knockout mice the host arginase inhibitor S-(2-boronoethyl)-L-cysteine (BEC), while inhibiting > 50% of the host arginase I activity in several tissues, did not block the ability of the rocF mutant H. pylori to colonize. In contrast, BEC poorly inhibited H. pylori arginase activity.
Conclusion: The in vivo source for the essential urea utilized by H. pylori urease is neither bacterial arginase nor host arginase II; instead, either residual host arginase I or agmatinase is probably responsible.
Keywords: Arginase; Helicobacter pylori; Mice; S-(2-boronoethyl)-L-cysteine; Urease.
Figures

Similar articles
-
Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity.J Bacteriol. 1999 Dec;181(23):7314-22. doi: 10.1128/JB.181.23.7314-7322.1999. J Bacteriol. 1999. PMID: 10572136 Free PMC article.
-
Purification and characterization of Helicobacter pylori arginase, RocF: unique features among the arginase superfamily.Eur J Biochem. 2004 May;271(10):1952-62. doi: 10.1111/j.1432-1033.2004.04105.x. Eur J Biochem. 2004. PMID: 15128304
-
Genetic microheterogeneity and phenotypic variation of Helicobacter pylori arginase in clinical isolates.BMC Microbiol. 2007 Apr 4;7:26. doi: 10.1186/1471-2180-7-26. BMC Microbiol. 2007. PMID: 17408487 Free PMC article.
-
Diversity, properties and functions of bacterial arginases.FEMS Microbiol Rev. 2021 Nov 23;45(6):fuab034. doi: 10.1093/femsre/fuab034. FEMS Microbiol Rev. 2021. PMID: 34160574 Review.
-
Unraveling the factors and mechanism involved in persistence: Host-pathogen interactions in Helicobacter pylori.J Cell Biochem. 2019 Nov;120(11):18572-18587. doi: 10.1002/jcb.29201. Epub 2019 Jun 25. J Cell Biochem. 2019. PMID: 31237031 Review.
References
-
- Blaser MJ. Gastric Campylobacter-like organisms, gastritis, and peptic ulcer disease. Gastroenterology. 1987;93:371–383. - PubMed
-
- Marshall BJ, Armstrong JA, McGechie DB, Glancy RJ. Attempt to fulfil Koch’s postulates for pyloric Campylobacter. Med J Aust. 1985;142:436–439. - PubMed
-
- Buck GE, Gourley WK, Lee WK, Subramanyam K, Latimer JM, DiNuzzo AR. Relation of Campylobacter pyloridis to gastritis and peptic ulcer. J Infect Dis. 1986;153:664–669. - PubMed
-
- Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–1131. - PubMed
-
- McGee DJ, Mobley HL. Mechanisms of Helicobacter pylori infection: bacterial factors. Curr Top Microbiol Immunol. 1999;241:155–180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
