

A comprehensive map of mobile element insertion polymorphisms in humans
- PMID: 21876680
- PMCID: PMC3158055
- DOI: 10.1371/journal.pgen.1002236
A comprehensive map of mobile element insertion polymorphisms in humans
Abstract
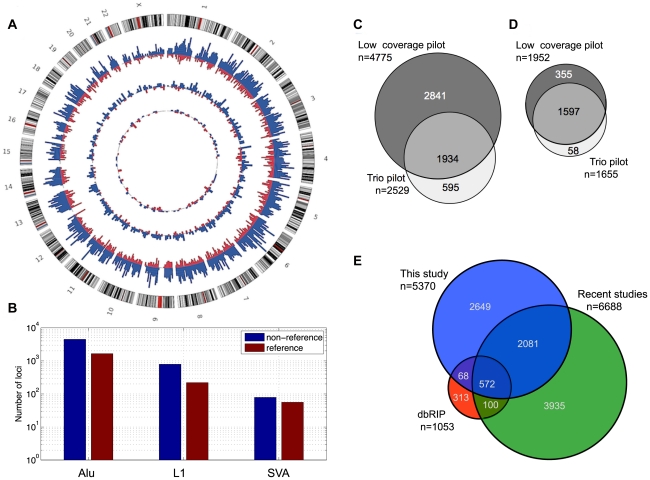
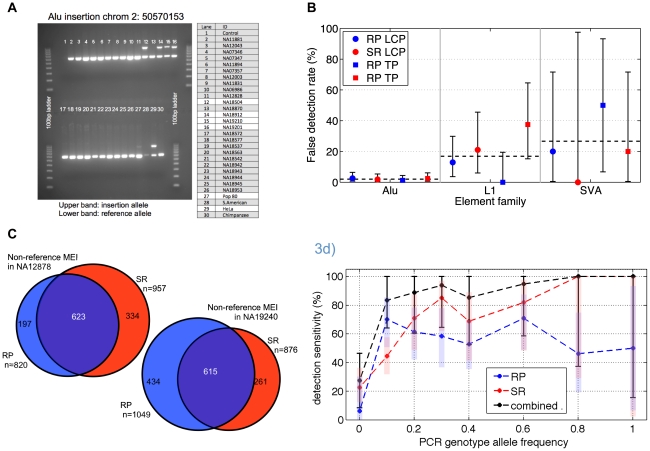
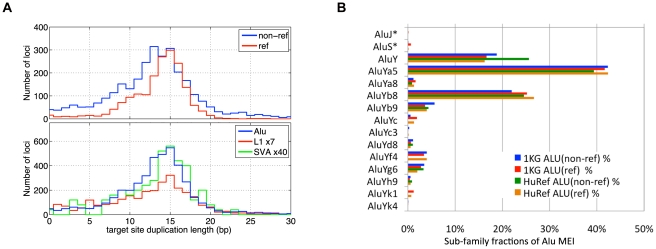
As a consequence of the accumulation of insertion events over evolutionary time, mobile elements now comprise nearly half of the human genome. The Alu, L1, and SVA mobile element families are still duplicating, generating variation between individual genomes. Mobile element insertions (MEI) have been identified as causes for genetic diseases, including hemophilia, neurofibromatosis, and various cancers. Here we present a comprehensive map of 7,380 MEI polymorphisms from the 1000 Genomes Project whole-genome sequencing data of 185 samples in three major populations detected with two detection methods. This catalog enables us to systematically study mutation rates, population segregation, genomic distribution, and functional properties of MEI polymorphisms and to compare MEI to SNP variation from the same individuals. Population allele frequencies of MEI and SNPs are described, broadly, by the same neutral ancestral processes despite vastly different mutation mechanisms and rates, except in coding regions where MEI are virtually absent, presumably due to strong negative selection. A direct comparison of MEI and SNP diversity levels suggests a differential mobile element insertion rate among populations.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




References
-
- Cordaux R, Hedges DJ, Batzer MA. Retrotransposition of Alu elements: how many sources? Trends Genet. 2004;20:464–467. - PubMed
-
- Deininger PL, Batzer MA, Hutchison CA, 3rd, Edgell MH. Master genes in mammalian repetitive DNA amplification. Trends Genet. 1992;8:307–311. - PubMed
-
- Mills RE, Bennett EA, Iskow RC, Devine SE. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183–191. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
